概要
1975年,日本大阪大学应用化学系工学院 (大阪大学工学部応用精密化学科, Department of Applied Chemistry, Faculty of Engineering, Osaka University)的村桥俊一 (村橋 俊一, Murahashi Shun-ichi)研究室报道了钯催化剂采用的卤代烯烃与烷基锂试剂进行的交叉偶联反应,研究发现反应具有优良的立体选择性,卤代烯烃构型完全保持[1]。

1979年,Murahashi进一步将锂试剂扩展至芳基与杂芳基锂,并将底物扩展至与芳基共轭的卤代烯[2],同样具有优良的立体选择性。

1982年,G. Linstrumelle将该反应条件应用于联烯基锂,成功实现联烯基锂的乙烯基化与芳基化[3]-[4]。

1987年,Pelter成功将Murahashi的反应条件扩展至苄基与肉桂基卤代烷[5],并获得良好的产率。

虽然上述反应条件具有广泛的底物适用范围与较高的产率。然而,部分底物在该条件下反应较慢,需要较高的温度、过量的有机锂试剂及较长的反应时间。同时,由于锂试剂较高的反应活性,反应过程中还会出现卤代烃之间的同偶联副反应及锂-卤素交换过程快于交叉偶联反应的问题。因此,无法适应大规模的实验室合成研究及工业生产。2010年,Yoshida课题组采用流动型微反应器 (flow microreactor)将Li-Br交换与Murahashi偶联成功结合,顺利完成了两种不同芳卤间的交叉偶联,获得较高的产率,并使反应时间极大缩短[6]。同时,有效避免了同种芳卤间的同偶联反应。

2013年,Feringa采用双(三叔丁基膦)合钯(II)或Pd-PEPPSI-IPent、Pd2(dba)3/三叔丁基膦及Pd2(dba)3/XPhos催化剂,成功将底物范围扩展至更多的芳卤及稠环芳卤,反应条件温和,反应时间有效缩短,同样避免了锂-卤素交换及同偶联等副反应。具有更为广泛的底物适用范围及优良的反应选择性与官能团兼容性,甚至连接有容易与锂试剂发生亲核反应的酮羰基、氰基及烷氧羰基官能团相关的底物,均可在该条件下顺利完成相应的交叉偶联[7]-[8]。

之后,Mori采用Ni(II)-NHC催化剂,实现了Murahashi偶联聚合 (Murahashicoupling polymerization),将该反应成功应用于π-共轭聚合物 (π-conjugatedpolymers)的合成[9]。

2019年,Feringa与Organ在深冷温度下 (cryogenic temperatures)下,采用反应活性更高的Pd-NHC催化剂,通过温度依赖的化学选择性引发策略 (temperature-dependent chemoselectivity trigger),成功实现氯代芳基溴 (芳基氯在该条件下无法参与偶联过程)的化学选择性交叉偶联[10]。同时,研究发现含有芳基氯官能团的偶联产物可以在一锅条件下完成多种后续的二级偶联过程,如Suzuki-Miyaura反应、Negishi反应及Buchwald-Hartwig反应等。

文献中将上述钯催化的卤代烃与有机锂试剂之间的交叉偶联反应称为Murahashi偶联反应 (Murahashi coupling)或Murahashi交叉偶联反应 (Murahashi cross-coupling)。该反应具有广泛的底物适用范围,优良的化学选择性与立体选择性及官能团兼容性。目前,Murahashi偶联反应在有机合成方法学研究、医药及有机材料研究领域具有极为广泛的应用前景。
基本文献
- [1] M. Yamamura, I. Moritani, S. Murahashi, J. Organomet. Chem. 1975, 91, C39. doi: 10.1016/S0022-328X(00)89636-9.
- [2] S. Murahashi, M. Yamamura, K. Yanagisawa, N. Mita, K. Kondo, J. Org. Chem. 1979, 44, 2408. doi: 10.1021/jo01328a016.
- [3] T. Jeffery-Luong, G. Linstrumelle, Synthesis 1982, 738. doi: 10.1055/s-1982-29923.
- [4] S. Murahashi, J. Organomet. Chem. 2002, 653, 27. doi: 10.1016/S0022-328X(02)01167-1.
- [5] A. Pelter, M. Rowlands, G. Clements, Synthesis 1987, 51. doi: 10.1055/s-1987-27840.
- [6] A. Nagaki, A.Kenmoku, Y. Moriwaki, A. Hayashi, J. Yoshida, Angew. Chem. Int. Ed. 2010, 49, 7543. doi:10.1002/anie.201002763.
- [7] M. Giannerini, M. Fañanás-Mastral, B. L. Feringa, Nat. Chem. 2013, 5, 667. doi: 10.1038/nchem.1678.
- [8] V. Hornillos, M. Giannerini, C. Vila, M. Fañanás-Mastral, B. L. Feringa, Org. Lett. 2013, 15, 5114. doi: 10.1021/ol402408v.
- [9] K. Fuji, S. Tamba, K. Shono, A. Sugie, A. Mori, J. Am. Chem. Soc. 2013, 135, 12208. doi: 10.1021/ja406374t.
- [10] N. Sinha, D. Heijnen, B. L. Feringa, M. G. Organ, Chem. Eur. J. 2019, 25, 9180. doi: 10.1002/chem.201901678.
反应机理
烯基卤参与的Murahashi偶联

芳卤参与的Murahashi偶联

参考文献
- [1] S. Murahashi, M. Yamamura, K. Yanagisawa, N. Mita, K. Kondo, J. Org. Chem. 1979, 44, 2408. doi:10.1021/jo01328a016.
- [2] M. K. Loar, J. K. Stille, J. Am. Chem. Soc. 1981, 103, 4174. doi: 10.1021/ja00404a033.
- [3] C. Amatore, A. Jutland, A. Suarez, J. Am. Chem. Soc. 1993, 115, 9531. doi: 10.1021/ja00074a018.
- [4] C. Amatore, M. Azzabi, A. Jutland, J. Am. Chem. Soc. 1991, 113, 8375. doi: 10.1021/ja00022a026.
反应实例
S1P5选择性拮抗剂关键中间体的合成[1]

联芳的合成[2]

Murahashi偶联聚合[3]

实验步骤
在氩气气氛下,向干燥的Schlenk瓶内加入Pd(PtBu3)2 (0.05 eq.)与卤代烃 (1 eq.),之后加入无水甲苯溶解 (维持底物浓度为0.15 M)。再将有机锂试剂 (1.2 eq.)加入适量溶剂稀释 (烷基锂试剂,采用无水甲苯稀释至浓度为0.36 M;烷基锂试剂,则采用无水THF稀释至浓度为0.6 M)。随后,通过注射泵缓慢加入 (超过1h后,滴加完毕)。滴加结束后,加入饱和NH4Cl溶液淬灭反应。淬灭完成后,将上述反应液加入乙醚进行萃取,合并有机相,减压除去溶剂。粗产物采用硅胶柱色谱 (根据不同产物,选择正戊烷或正戊烷/乙酸乙酯100:3作为洗脱剂)进行分离纯化,获得最终目标产物。
参考文献
- [1] M. Giannerini, M. Fañanás-Mastral, B. L. Feringa, Nat. Chem. 2013, 5, 667. doi: 10.1038/nchem.1678.
- [2] V. Hornillos, M. Giannerini, C. Vila, M. Fañanás-Mastral, B. L. Feringa, Org. Lett. 2013, 15, 5114. doi: 10.1021/ol402408v.
- [3] K. Fuji, S. Tamba, K. Shono, A. Sugie, A. Mori, J. Am. Chem. Soc. 2013, 135, 12208. doi: 10.1021/ja406374t.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Yamamoto偶联 (Yamamoto coupling)又称为Yamamoto聚合 (Yamamoto polymerization),是过渡金属试剂 (如PdCl2(bipy), NiCl2(bipy), Ni (cod)2,NiBr2(PPh3)2, NiCl2, CoCl2, FeCl2等,其中NiCl2(bipy)与Ni (cod)2最为常用)促进的二卤代芳烃与多卤代芳烃通过去卤化反应而进行偶联缩聚[1]或脱卤C-C偶联 (dehalogenative carbon-carbon coupling reactions)[2]过程,获得聚芳烃类聚合物[1]及环低聚 (环三聚最为常见)产物[2]的反应。该反应在1978年由日本东京工业大学资源化学研究所(東京工業大学資源化学研究所,Research Laboratory of Resources Utilization, Tokyo Institute of Technology)的山本隆一 (山本 隆一,Yamamoto Takakazu)研究室首次报道[1]-[2]。

之后,Yamamoto将上述反应条件进行了深入研究,并成功应用于聚吡啶[3]-[4]、聚啉[5]与聚异喹啉[5]、聚噻吩[6]、吡啶-硒吩共聚物[7]、聚氢醌[8]、聚对苯醌[8]与聚对二乙酰氧基苯单元[8]、光学活性[9]及氧化还原活性聚合物[9]的合成。


目前,该反应已经广泛应用于具有较高聚合度与热稳定性的各类π-共轭聚合物 (π-conjugated polymer) [1]-[10]、共轭微孔聚合物[11] (Conjugated Microporous Polymer, CMP)及全芳香梯形聚合物[12]等的合成。
基本文献
- [1] T. Yamamoto, Y. Hayashi, A. Yamamoto, Bull. Chem. Soc. Jpn. 1978, 51, 2091. doi: 10.1246/bcsj.51.2091.
- [2] Z. Zhou, T. Yamamoto, J. Organomet. Chem. 1991, 414, 119. doi: 10.1016/0022-328X(91)83247-2.
- [3] T. Yamamoto,T. Ito, K. Kubota, Chem.Lett. 1988, 17, 153. doi: 10.1246/cl.1988.153.
- [4] T. Maruyama, K. Kubota, T. Yamamoto, Macromolecules 1993, 26, 4055. doi: 10.1021/ma00067a052.
- [5] T. Kanbara, N. Saito, T. Yamamoto, K. Kubota, Macromolecules 1991, 24, 5883.
- doi: 10.1021/ma00021a027.
- [6] T. Yamamoto, A. Morita, Y. Miyazaki, T. Maruyama, H. Wakayama, Z. Zhou, Y.
- Nakamura, T. Kanabara, Macromolecules 1992, 25, 1214. doi: 10.1021/ma00030a003
- [7] T. Yamamoto, N. Hayashida, React. Funct. Polym. 1998, 37, 1. doi: 10.1016/S1381-5148(97)00140-5.
- [8] T. Yamamoto, T. Kimura, K. Shiraishi, Macromolecules 1999, 32, 8886. doi: 10.1021/ma9907946.
- [9] U. Scherf, E. J. W. List, Adv. Mater. 2002, 14, 477. doi: 10.1002/1521-4095(20020404)14:7<477::AID-ADMA477>3.0.CO;2-9.
- [10] T. Yamamoto, J. Synth. Org. Chem. Jpn.1995, 53, 999. doi: 10.5059/yukigoseikyokaishi.53.999.
- [11] J. Schmidt, M. Werner, A. Thomas, Macromolecules 2009, 42, 4426. doi: 10.1021/ma9005473.
- [12] K. Chmil, U. Scherf, Makromol. Chem. Rapid Commun. 1993, 14, 217. doi: 10.1002/marc.1993.030140401.
反应机理
低聚反应

聚合反应

参考文献
- [1] T. Yamamoto, S. Wakabayashi, K. Osakada, J. Organomet. Chem. 1992, 428, 223. doi: 10.1016/0022-328X(92)83232-7.
- [2] T. Yamamoto, T. Koizumi, Polymer, 2007, 48, 5449. doi: 10.1016/j.polymer.2007.07.051.
- [3] T. Kohara, T. Yamamoto, A. Yamamoto, J. Organomet. Chem. 1980, 192, 265. doi: 10.1016/S0022-328X(00)94431-0.
- [4] S. Komiya, Y. Abe, A. Yamamoto, T. Yamamoto, Organometallics 1983, 2, 1466. doi: 10.1021/om50004a042.
- [5] K. Tatsumi, A. Nakamura, S. Komiya, T. Yamamoto, A. Yamamoto, J. Am. Chem.Soc. 1984, 106, 8181. doi: 10.1021/ja00338a029
- [6] D. R. Fahey, J. E. Mahan, J. Am. Chem.Soc. 1977, 99, 2501. doi: 10.1021/ja00450a017.
- [7] D. R. Fahey. J. Am. Chem. Soc. 1970, 92, 402. doi: 10.1021/ja00705a612
- [8] F. Ozawa, T. Hidaka, T. Yamamoto, A. Yamamoto, J. Organomet. Chem. 1987, 330, 253. 10.1016/0022-328X(87)80292-9.
反应实例
联芳的合成[1]

聚噻吩的合成[2]

手性聚合物的合成[3]-[4]

多孔共价有机聚合物 (porous covalent organic polymers, COPs)的合成[5]

取代三萘的合成[6]


炔基取代Starphenes的合成[8]

共轭微孔聚合物 (Conjugated Microporous Polymer, CMP)的合成[9]

Benzophenes的合成[10]

全芳香梯形聚合物的合成[11]

实验步骤
聚合反应
在手套箱内,向管式反应瓶 (tubular reaction vessel)中加入Ni(COD)2 (1.45 eq.)、2,2′-联吡啶(1.93 eq.)、芳基卤 (1 eq.)、cis-1,5-环辛二烯 (5.72 eq.)及无水THF (维持底物浓度为0.15 M)。将反应瓶在手套箱内密封后,在微波辐射下加热 (温度为120°C) 12 min。减压除去溶剂后,将残余物溶于煮沸的CHCl3中,并通过短硅胶柱过滤,除去难溶性盐。 粗产物通过三次连续研磨粉碎 (successive trituration)的方式进行纯化,固体通过离心机分离进行回收: 第一次在EtOH中回流45 min,第二次在乙腈中,室温下超声30 min,最后在甲苯/乙醇中回流45 min,获得最终目标产物。
低聚反应
室温下,向Ni(cod)2 (1.3 eq.)的DMF溶液 (浓度为0.43 M)中加入1,5-环辛二烯 (1.6 eq.)与2,2′-联吡啶 (1.3 eq.). 随后,滴加芳基卤 (1 eq.)的DMF溶液 (浓度为0.52 M)。将上述反应混合物在70oC下搅拌22h后,加入适量水及乙醚进行萃取。将乙醚相采用无水硫酸镁进行干燥,过滤。减压除去溶剂后,将残余物采用硅胶柱色谱分离纯化,再用乙醚进行重结晶,获得环三聚产物。
参考文献
- [1] Z. Zhou, T. Yamamoto, J. Organomet. Chem. 1991, 414, 119. doi: 10.1016/0022-328X(91)83247-2.
- [2] T. Yamamoto, M. Omote, Y. Miyazaki, A. Kashiwazaki, B. Lee, T. Kanbara, K. Osakada, T. Inoue, K. Kubota, Macromolecules 1997, 30, 7158. doi: 10.1021/ma9708104.
- [3] T. Yamamoto, T. Asao, H. Fukumoto, Polymer, 2004, 45, 8085. doi: 10.1016/j.polymer.2004.09.060.
- [4] T. Ijima, T. Yamamoto, Chem. Lett., 2005, 1672. doi: 10.1246/cl.2005.1672.
- [5] L. Guo, D. Cao, J. Mater. Chem. C, 2015, 3, 8490. doi: 10.1039/C5TC01649E.
- [6] E. C. Rüdiger, F. Rominger, L. Steuer, U. H. F. Bunz, J. Org. Chem. 2016, 81, 193. doi: 10.1021/acs.joc.5b02481.
- [7] V. Berezhnaia, M. Roy, N. Vanthuyne, M. Villa, J.-V. Naubron, J. Rodriguez, Y. Coquerel, M. Gingras, J. Am. Chem. Soc. 2017, 139, 18508. doi: 10.1021/jacs.7b07622.
- [8] E. C. Rüdiger, M. Porz, M. Schaffroth, F. Rominger, U. H. F. Bunz, Chem. Eur. J. 2014, 20, 12725. doi: 10.1002/chem.201403697.
- [9] J. Schmidt, M. Werner, A. Thomas, Macromolecules 2009, 42, 4426. doi: 10.1021/ma9005473.
- [10] E. C. Rüdiger, S. Koser, F. Rominger, J. Freudenberg, U. H. F. Bunz, Chem.Eur. J. 2018, 24, 9919 , doi: 10.1002/chem.201801459.
- [11] K. Chmil, U. Scherf, Makromol. Chem. Rapid Commun. 1993, 14, 217. doi: 10.1002/marc.1993.030140401.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者:alberto-caeiro
Liebeskind–Srogl偶联反应是过渡金属铜和钯协同催化的硫酯和硼酸间的C-C偶联成键反应[1]。该反应是由捷克科学院Jiri Srogl教授和美国埃默里大学Lanny S. Liebeskind教授共同发现。如下图所示,该反应中的硼酸可替换成其他亲核试剂,如:锡试剂,硅试剂,锌试剂,格氏试剂,以及炔烃和胺等;而另一部分可替换成硫酯,环状硫酰胺,芳基硫醚和炔基硫醚等[2]。

反应机理如下图所示:底物中的硫与铜配位活化C-S键后,金属钯对C-S键氧化加成,硼酸转金属离去铜Cu-S物种,随后二价钯物种还原消除得到C-C键偶联产物并再生催化剂零价钯,因此该反应中需要催化量的钯,当量的铜作为催化剂。

反应发展
Jiri Srogl教授和Lanny S. Liebeskind教授最开始发现该反应时需要当量的铜作为共催化剂,金属钯作用催化活性物种在无氧气的条件下进行,其机理如前所述。
作为还反应的改进版, Jiri Srogl教授和Lanny S. Liebeskind教授于2007年报道了无需金属钯,催化量的铜在氧气的条件下,与2.5当量硼酸即可实现此反应,并推测此反应可能经过高价铜中间体[3]。2.5当量的硼酸除了作为偶联片段,还作为Lewis酸再生铜催化剂。

2009年,Lanny S. Liebeskind教授通过在底物中引入N-O键以模仿金属硫蛋白体系中的S-S键,从而实现Cu-S键的断裂,实现铜催化剂的再生,减少硼酸的用量[4]。该反应是在微波条件下进行。在假设的反应机理中,R2可直接与羰基部分结合得到产物和Cu-S键,随后在微波和高温作用下,Cu-S键断裂再生催化剂,无需另一当量的硼酸。


应用实例
特拉华大学的Joseph M. Fox教授近期在JACS上发表了金作为协同催化剂的Liebeskind-Srogl Coupling反应{5},实现了四嗪片段的快速高效引入。反应具有良好的底物适用范围,并有很高的收率,为复杂分子中引入四嗪提供了新的方法。

参考文献
- [1] For a seminal work, see: Liebeskind, L.; Srogl, Jiri, J. Am. Chem. Soc. 2000, 122: 11260. DOI: 10.1021/ja005613q; For recent reviews, see: a, Hana Prokopcovμ,; C. Oliver Kappe, Angew. Chem. Int. Ed. 2009, 48, 2276. DOI: 10.1002/anie.200802842; b, Sachin G. Modha,; Vaibhav P. Mehta,; Erik V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 5042. DOI: 10.1039/C3CS60041F.
- [2] Hong-Gang Cheng,; Han Chen,; Yue Liu,; Qianghui Zhou. Asian J. Org. Chem. 2018, 7, 490. DOI: 10.1002/ajoc.201700651.
- [3] Janette M. Villalobos,; Jiri Srogl,; Lanny S. Liebeskind, J. Am. Chem. Soc., 2007, 129, 15734. DOI: 10.1021/ja074931n.
- [4] Zhihui Zhang,; Matthew G. Lindale,; Lanny S. Liebeskind. J. Am. Chem. Soc., 2011, 133, 6403. DOI: 10.1021/ja200792m.
- [5] William Lambert, Yinzhi Fang, Subham Mahapatra, Zhen Huang, Christopher W. am Ende, Joseph M. Fox, J. Am. Chem. Soc., Just Accepted Manuscript • DOI: 10.1021/jacs.9b08677.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者 孙苏赟
-
有机锌试剂的偶联反应和其他金属有机化合物
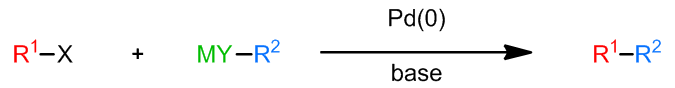
在这类反应中,X为卤素,一般为碘,溴和三氟磺酸酯,氯的反应太慢了,但是不免有一些例子;R1通常是不饱和的芳基,烯基,也有一些苄基和烯丙基的例子;R2的选择性也很广,可以使烷基、烯基、芳基、苄基、烯丙基和炔基;M若是Zn,则是Negishi反应,Mg对应Kumada-Corriu反应,Cu和其他一些金属也是可以的;催化剂方面,Pd(0/II)和Ni(0)都可以使用。
机理和Stille反应、Suzuki反应是类似的。几个例子:
(1)sp2-sp3中心的偶联:ref.1
这是Negish改进的反应条件,乙烯基溴类衍生物是不可以发生类似的反应的因为活性不够。
(2)三幅磺酸酯的Kumada-Curriu偶联:ref.2
(3)氯代烃偶联的例子:ref.3
(4)有机锆试剂-Negishi串联反应:ref.4
(5)Negishi反应用于合成非天然的氨基酸:ref.5
(6)铁催化的Kumada-Corriu偶联:ref.6
-
炔铜化合物的偶联:Sonogashira偶联
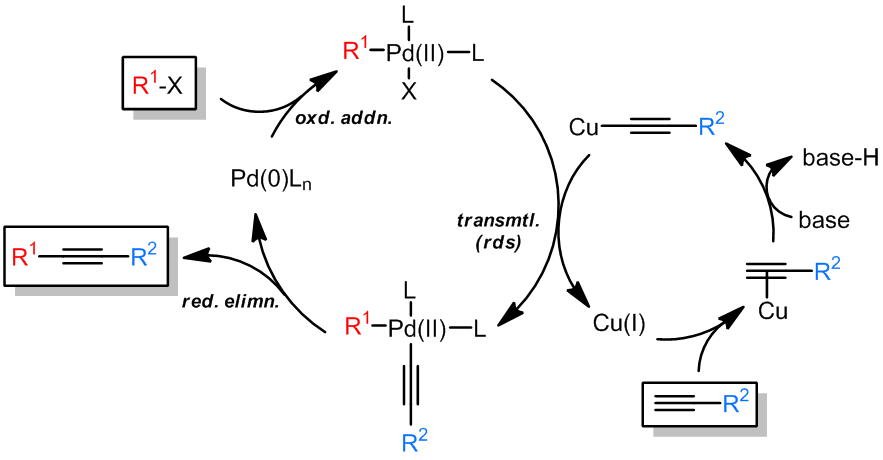
在偶联反应中,有一些反应是进行sp-C的偶联的,也就是炔化合物的反应。虽然炔基的硼试剂、锌试剂和锡试剂在上述的反应中都非常有用,但是最常见的炔的偶联还是Sonogashira反应,最早的研究是Castro-Sterphens进行的化学计量的炔基铜在吡啶中回流进行的,金属Pd的使用是的反应的条件变得温和且高效:
反应中,X为卤素,一般为碘,溴和三幅磺酸酯,R1是芳基,烯基;R2可以是烷基、烯基、芳基、苄基、烯丙基和炔基;但是Cu(I)是不可缺少的。
- 反应机理
- 两个例子

(1)区域选择的偶联反应:ref.7
(2)串联的脱保护-Sonogashira偶联:ref.8
REFERENCES
- J. Am. Chem. Soc., 2000, 122 (36), pp 8654–8664, DOI: 10.1021/ja0015287
- J. Am. Chem. Soc., 2000, 122 (16), pp 3811–3820, DOI: 10.1021/ja9939439
- Org. Lett., 2001, 3 (15), pp 2419–2421, DOI: 10.1021/ol016274o
- J. Am. Chem. Soc., 1999, 121 (2), pp 456–457, DOI: 10.1021/ja983429n
- J. Am. Chem. Soc., 2012, 134 (22), pp 9291–9295, DOI: 10.1021/ja301326k
- J. Am. Chem. Soc., 2004, 126 (12), pp 3686–3687, DOI: 10.1021/ja049744t
- J. Am. Chem. Soc., 2000, 122 (23), pp 5473–5476, DOI: 10.1021/ja0007197
- J. Am. Chem. Soc., 2002, 124 (51), pp 15196–15197, DOI: 10.1021/ja028936q
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
某些烷基镁试剂,锌试剂和锡试剂都可以参与欧联反应,但是有机硼试剂比较特殊,它特殊在于烷基硼试剂可以通过烯烃的硼氢化反应直接得到。反应的催化剂和碱的使用因此是更加关键的变量,特别是乙烯基硼试剂和芳基硼试剂的反应。通常来讲,PdCl2(dppf),NaOH水溶液和THF作为溶剂是成功率比较高的反应条件,反应机理也和之前的描述相同。
在这些偶联反应中,只会发生初级烷基的迁移反应,二级烷基很难迁移,反应速率明显低于一级烷基,因此经常使用的是没有一级烷基的硼氢化试剂,例如9-BBN,Sia2BH,等:
此外,三氟硼酸盐也可以达到很好的Suzuki偶联的效果。
一些例子:
(1)9-BBN的使用:ref. 1
(2)分子内的偶联:ref. 2
(3)氟芳烃的衍生物作为底物的偶联:ref. 3
(4)通过锂试剂得到的硼试剂:ref. 4
在这个例子中,烷基硼不是烯烃由硼氢化反应得到的,而是由锂试剂进行类似转金属化的反应得到的,而实际操作是利用MeO-9-BBN淬灭具有活性的烷基锂中间体的。
(5)硼酸环丙烷衍生物的偶联:ref. 5
硼酸环丙烷衍生物是一个很好的sp3-C中心,最后的产物也是具有光学活性的。
(6)ref. 6
五、Suzuki-Miyaura反应的另外一些例子
- Suzuki反应的Johnson改进: ref. 7,8
- 偕二溴代烯烃区域选择的Suzuki偶联:ref. 9
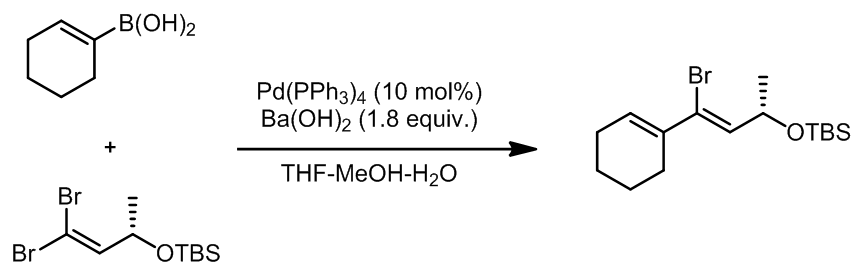
- 季铵碱的Suzuki偶联:ref. 10
- 烯炔发生Suzuki偶联 ref. 11




烯炔中的双键可以区域选择的进行硼氢化反应得到的硼烷化合物,因而进行Suzuki偶联。
- 烯醇磺酸酯的Suzuki偶联:ref. 12


References
- J. Am. Chem. Soc., 1991, 113 (14), pp 5402–5410, DOI: 10.1021/ja00014a036
- Tetrahedron Lett., 1992, 33, 2571, DOI: 1016/S0040-4039(00)92245-7
- Org. Lett., 2011, 13 (15), pp 3956–3959, DOI: 10.1021/ol2014768
- J. Org. Chem., 1998, 63 (22), pp 7885–7892, DOI: 10.1021/jo9811423
- Angew Chem Int Ed., 1998, 37, 2845,DOI 10.1002/(SICI)1521-3773(19981102)37:20<2845::AID-ANIE2845>3.0.CO;2-U
- J. Am. Chem. Soc., 2001, 123 (41), pp 10099–10100, DOI: 10.1021/ja011306o
- Angew Chem Int Ed., 2002, 3192,DOI 10.1002/1521-3773(20020902)41:17<3192::AID-ANIE3192>3.0.CO;2-E
- Org. Synth., 1997, 75, 69, DOI: 10.15227/orgsyn.75.69
- Org. Lett., 2002, 4 (11), pp 1955–1957, DOI: 10.1021/ol0259746
- J. Am. Chem. Soc., 2003, 125 (17), pp 5040–5050, DOI: 10.1021/ja029216m
- J. Org. Chem., 1990, 55 (19), pp 5324–5335, DOI: 10.1021/jo00306a007
- Org. Lett., 2012, 14 (12), pp 3186–3189, DOI: 10.1021/ol301278e
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者 孙苏赟
 X为卤素,一般为溴或是碘,也可能是三氟甲基磺酸盐,但是也有一些例子中会使用氯代烃参与反应;R1通常是不饱和的芳基,烯基,但是苄基和烯丙基在某些例子中也可以;R2的选择性也很广,可以使烷基、烯基、芳基、苄基、烯丙基和炔基,而对于R’的选择,一般使用烷氧基(硼酸酯)或是羟基(硼酸),也有使用烷基的例子,例如:
X为卤素,一般为溴或是碘,也可能是三氟甲基磺酸盐,但是也有一些例子中会使用氯代烃参与反应;R1通常是不饱和的芳基,烯基,但是苄基和烯丙基在某些例子中也可以;R2的选择性也很广,可以使烷基、烯基、芳基、苄基、烯丙基和炔基,而对于R’的选择,一般使用烷氧基(硼酸酯)或是羟基(硼酸),也有使用烷基的例子,例如:
或者将以上的三个例子换成对应的硼酸酯或三氟硼酸钾 (-BF3K)。需要注意的是有一类不可参与Suzuki偶联的硼试剂的特殊例子:
此外,反应中需要碱的参与,一促进发生还原消除反应,可使用的碱可以使碳酸盐,碱金属氢氧化物类,膦类,胺类,甚至是F–也可以作为碱使用;至于核心的催化剂方面,Pd(0)和Pd(II)都有很多可行的例子,Ni(0)也可以作为反应的催化剂。
Suzuki反应是一个基于硼试剂的过渡金属催化偶联反应,和Stille反应非常相似。令人欣慰的是,和Stille反应中使用的锡试剂相比,例如硼酸类化合物的硼试剂的毒性大都很低,因而Suzuki反应在实际的合成过程中更加频繁的使用,用于形成新的碳碳键。同时,反应的条件大都十分温和,对于实际操作的要求不是非常高,反应的溶剂有很多种选择,甚至有一些在水中反应的例子。
- 反应机理
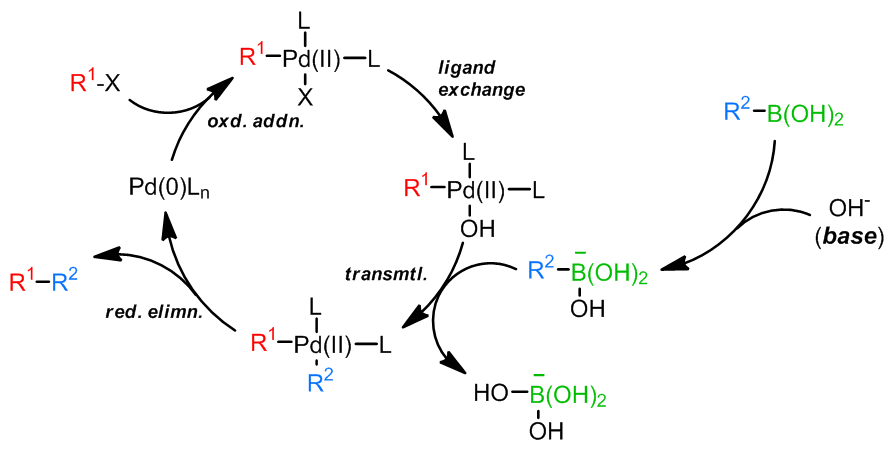

对于乙烯基类的硼试剂,反应中底物和产物的双键构型是一致的,并且Suzuki反应具有很好的官能团兼容性,碱和催化剂种类选择范围很广,因此在实际中的应用很多很广。
和Stille偶联一样,关于反应机理中转金属化的过程是否会产生 Pd(IV)还是一个具有争议的问题,因为 Pd(IV)对于Pd来说是一个非常不常见的氧化态;此外,在大多数过程中,会有一个四面体硼酸中间体产生,这个中间体的产生对于转金属化步骤是很有利的,可以加快转金属化;普遍认为氧化加成步骤是最慢的步骤,因此造成了它为Sukuzi偶联的决速步骤。烷基-BBN参与的反应中不会发生β-消除,因为还原消除太快了。另外一点,如果反应是Ni(0)催化,那么反应机理会产生自由基中间体。
- 硼试剂的制备
i. 炔的硼氢化反应
ii. 金属有机试剂和硼酸酯反应
iii. 双硼试剂的Miyaura偶联 ref. 1
实际上这也是一种Suzuki偶联,许多种双硼试剂现在都是商业化的试剂,这个途径非常方便。这里有一个实际应用:ref. 2
iv. 直接法C-H活化硼化反应 ref. 3,4
3. 芳基硼酸(酯)和烯基硼酸(酯)的交叉偶联
(1)邻位取代的芳基硼酸衍生物的生成和偶联反应:ref.5
(2)烯基硼试剂的例子:ref.6
在所有这类偶联反应中,所有双键的构型是不会发生改变的。这个反应中使用了TlOH,这是一种非常高效的碱,可以极大的加速Suzuki偶联过程。
(3)偕二溴代烃的区域选择性:ref.7
反应更加倾向于发生在位阻更小的卤原子上。
(4)一个固相反应的例子:ref.8
4.催化剂体系的发展
i. 芳基氯的偶联 ref. 9
相对于溴和碘来说,芳基氯代烃更加受到青睐,但是大多时候它们在Suzuki反应表现出的是惰性的,因此有很多种含磷的配体被发明出来,以提高芳基氯在Suzuki欧联中的反应活性。这个反应的条件中使用的tBu3P是促使这个反应成功的关键,碰巧的是这个反应条件对于Stille偶联也是同样高效的。Ref. 10
ii. 联苯二烷基磷类配体
这一类配体是由Buchwald最早开发出来的,他们可以催化一些Suzuki偶联。这些配体是二联苯类衍生物,这些配体的位阻大都很大,他们在空气中很稳定,两个苯环中间的单键是不可以自由旋转的,他们的作用认为是加速了氧化加成的步骤,此外他们的大位阻非常高效的促进了单络合物种Pd(0)-PR3的形成:
烷基R1使得磷元素上电子密度很高,有利于氧化加成步骤的发生,也有利于中心金属 Pd(II)的稳定性;大体积的R2/3可以加速还原消除反应,并且提高了Pd元素的使用频率,可以使得活性中心为LPd(0)而不是L2Pd(0),并且使催化剂性质稳定。
这里是几个例子:
(1)大位阻的底物:ref.11
(2)NHC作为配体的例子:ref.12
(3)烷基溴/碘的例子:ref.13
这样的反应条件可以遏制Pd(II)的β-消除步骤。
(4)由炔转化得到共轭二烯:ref.14
(5)双硼化合物的单偶联:ref.15
(6)不对称的Suzuki偶联:ref.16
REFERENCES
- J. Org. Chem., 1995, 60 (23), pp 7508–7510, DOI: 10.1021/jo00128a024
- Angew Chem Int Ed., 2001, 40, 1967, DOI: 10.1002/1521-3773(20010518)40:10<1967::AID-ANIE1967>3.0.CO;2-Q
- Chem. Rev., 2010, 110 (2), pp 890–931, DOI: 10.1021/cr900206p
- J. Am. Chem. Soc., 2002, 124 (3), pp 390–391, DOI: 10.1021/ja0173019
- Tetrahedron Lett., 1985, 26, 5997, DOI: 1016/S0040-4039(00)95108-6
- J. Am. Chem. Soc., 1987, 109 (15), pp 4756–4758, DOI: 10.1021/ja00249a069
- J. Org. Chem., 1997, 62 (25), pp 8708–8721, DOI: 10.1021/jo970960c
- J. Org. Chem., 1999, 64 (11), pp 3885–3890, DOI: 10.1021/jo982135h
- Angew Chem Int Ed., 1998 37 3387, DOI:10.1002/(SICI)1521-3773(19981231)37:24<3387::AID-ANIE3387>3.0.CO;2-P
- Angew Chem Int Ed., 2002 41 4176,DOI:10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U
- Angew Chem Int Ed., 2004 43 1871 1002/anie.200353615
- Chem. Eur. Chem., 2006, 12, 4743, DOI: 10.1002/chem.200600251
- J. Am. Chem. Soc., 2004, 126 (5), pp 1340–1341, DOI: 10.1021/ja039889k
- Tetrahedron 1983, 39, 3271, DOI: 1016/S0040-4020(01)91575-3
- J. Am. Chem. Soc., 2008, 130 (2), pp 466–468, DOI: 10.1021/ja078129x
- J. Am. Chem. Soc., 2002, 124 (40), pp 12042–12053, DOI: 10.1021/ja020788g
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者 孙苏赟

X为卤素或是磺酸酯类;对于底物的选择性,R1可以是芳基,烯基,但是苄基和烯丙基也是可以的,R2对于底物活性:炔基 > 烯基 > 芳基 > 苄基 > 甲基 > 烷基;作为稀释剂来说,R’应该是一个转移性较差的基团,以使得R2可以顺利的和金属发生相互作用;反应中也可以添加Cu+,LiCl以加速反应的发生。
在Stille反应中,有很多种可以使用的溶剂,并且反应中不需要碱的参与!对于锡试剂来说,即使是四烷基锡,例如Me4Sn或Bu4Sn,在反应中都会向催化剂金属转移烷基,即使是在高温也是如此,并且Pd(II)几乎不发生β-消除,并且Stille反应对于底物中官能团的忍耐性也很好。值得注意的一点是锡试剂的毒性大都很高,这在制备和使用锡试剂的过程中需要格外注意,甚至是在反应后期的提纯和处理工作中也是如此,对于含有-SnMe3的化合物尤是如此,-SnBu3化合物则相对较安全。
Stille反应的介绍:
施蒂勒反应(Migita-Kosugi-Stille Cross Coupling)
- 烷基锡试剂的制备
i.炔的锡氢化反应 ref. 1
ii.金属有机物和锡卤化物的反应
其中,R就是需要转移并且可以发生转移的基团。
iii. Takai反应
Takai反应是从醛酮化合物制得双键的方法,这个反应我在《碳碳双键的形成》中有详细的描述,请参见:XXXXXXXXXXXXXXXXXXXXX
几个例子:
(1)Sp2-锂试剂的方法:Ref. 2
(2)炔丙基醇的反应:Ref. 3
(3)炔烃转化成为锡烷:Ref. 4
(4)一个钯催化的锡氢化反应:Ref. 5
这个反应中,因为三键的两侧链接的是不同的基团造成了选择性的差异。Joc 1990 1857
(5)另一个钯催化的反应:Ref. 6
(6)Pattenden在后期对此类的反应进行了改进:Ref. 7
这个方法中,产生的中间体使得第二步的区域选择性和立体选择性非常好,这个步骤叫做Pattenden的改进,产生的乙烯基溴可以在原位转化成锡烷化合物:
- 有机锡化物的偶联
有机锡试剂的偶联反应中,钯试剂作为催化剂也有着很广泛的应用。Farina对于配体在反应中的作用做了很深入的研究,得知三呋喃基磷 (tris(2-furyl)phosphine, TFP),或Ph3As类软碱化合物作为配体参与反应的效果最佳,因为他们可以加速中间体的转金属化过程。此外,碱,LiCl和CuI有时是反应中不可少的添加剂。总的来看,添加剂和配体在反应中的作用尚不十分清楚,反应的条件也没有较为统一的标准需要具体问题具体分析,需要根据实际情况进行调整。
- 反应机理
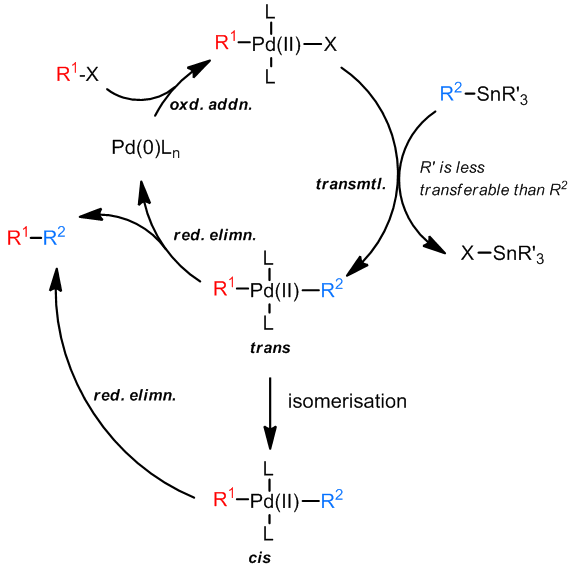

对于这个反应的过程中,一般认为转金属化过程是没有Pd(IV)中间体参与的,因为这个氧化态的Pd元素是很不常见的,从机理上也可以看出反应不需要碱来帮助发生还原消除步骤。
此外,CuCl可以加速Stille偶联,特别是对于大位阻的锡烷化合物,可能的原因是因为反应中发生了烷基锡到烷基铜的转金属化过程,一次来加速ArPd(II)X的转金属化过程。Ref. 9
- Stille偶联的应用
(1)羟基醇的反应:Ref. 10
底物中虽然有羟基,但是这是不需要进行保护的,它不会影响反应的结果,并且底物和产物中双键的构型是一致的。
(2)烯醇磺酸酯的偶联:Ref. 11
事实证明苯磺酸酯底物也是可以进行交叉偶联的,其中磺酸酯可以从酮经过烯醇变换再磺化得到。
(3)另一个双Stille偶联的反应:Ref. 12
(4)使用CuCl和DMSO作为溶剂在这个反应中是至关重要的,如果没有CuCl的加入,那么这个反应就不能发生: 13
(5)一个室温下的反应:Ref. 14
其中,配体也可是Cy3P,其他的配体也有测试,但是效果普遍不如这两种配体。
(6)使用了CdCl2加速转金属化:Ref. 15
(7)Pi-烯丙基的Stille偶联:Ref. 16
(8)Ref. 17
值得注意的是,磺酸酯在这个反应是不会受到影响的。
(9)烯丙基卤代烃的例子:Ref. 18
烯丙基卤代烃是一个参与Stille偶联很好的反应底物,这个条件中使用了Ph3As作为配体,也叫作Farina的改进条件。
(10)锡催化的Stille偶联:Ref. 8
这是一个一步法将端炔转化成偶联的烯烃产物的方法,是一个钯催化的锡氢化反应-Stille偶联串联反应,其中锡氢化合物可以进行循环催化,总的来看这个步骤中有94%的锡元素可以重复利用。
(11)酰氯的例子:
在Stille欧联中,酰氯也是一个备选的底物,反应的机理也是类似的,酰氯先经过氧化加成和Pd发生相互作用,之后发生分子间的偶联过程。此外,CO也同样可以参与类似的反应。
(12)碘代茂的反应:Ref. 2
(13)在rapamycin的全合成中的应用:(not published yet)
这个例子中,使用了两次Stille偶联,第一次是用于合成锡烷,第二次是用于偶联形成新的碳碳键。
REFERENCES
- Chem. Rev., 2000, 100 (8), pp 3257–3282, DOI: 10.1021/cr9902695
- Angew. Chem. Int. Ed. Engl., 1993, 1653, DOI:10.1002/anie.199316531
- Org. Lett., 1999, 1 (7), pp 1137–1139, DOI: 10.1021/ol990967b
- J. Am. Chem. Soc., 1998, 120 (47), pp 12237–12254, DOI: 10.1021/ja981846u
- J. Am. Chem. Soc., 1998, 120 (26), pp 6477–6487, DOI: 10.1021/ja980786p
- J. Am. Chem. Soc., 2000, 122 (16), pp 3830–3838, DOI: 10.1021/ja994285v
- J. Am. Chem. Soc., 2000, 122 (16), pp 3830–3838, DOI: 10.1021/ja994285v
- J. Am. Chem. Soc., 2001, 123 (14), pp 3194–3204, DOI: 10.1021/ja0035295
- J. Am. Chem. Soc., 1999, 121 (33), pp 7600–7605, DOI: 10.1021/ja991500z
- J. Am. Chem. Soc., 1987, 109 (3), pp 813–817, DOI: 10.1021/ja00237a029
- J. Am. Chem. Soc., 1984, 106 (16), pp 4630–4632, DOI: 10.1021/ja00328a063
- J. Org. Chem., 1997, 62 (24), pp 8290–8291, DOI: 10.1021/jo971793j
- J. Am. Chem. Soc., 1999, 121 (33), pp 7600–7605, DOI: 10.1021/ja991500z
- J. Am. Chem. Soc., 2003, 125 (13), pp 3718–3719, DOI: 10.1021/ja0344563
- J. Am. Chem. Soc., 2002, 124 (10), pp 2259–2262, DOI: 10.1021/ja011931t
- Org. Lett., 2002, 4 (12), pp 2063–2066, DOI: 10.1021/ol0259342
- J. Am. Chem. Soc., 2003, 125 (20), pp 6261–6271, DOI: 10.1021/ja034525d
- Synlett 1994; 1994(3): 181-182, DOI: 1055/s-1994-22785
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
课题的提出
最近几十年,金属催化主要用于C-H键的活化,构建新的C-C键。但该催化体系在催化交叉偶联反应时极易伴随自身偶联副产物的生成[1]。至今为止,还没有关于能完全抑制自身偶联副产物生成的报道。考虑到Fe催化C-H键活化过程中会与Lewis酸作用生成有机铁中间体III[2], 东京大学Eiichi Nakamura、尚睿团队猜想若底物带有碱性基团,则两个底物可通过碱性基团发生瞬态连接形成中间体II和III的平衡态,然后,III缓慢脱去质子可生成中间体IV,紧接着,IV发生交叉偶联反应,得到产物V(Figure 1)。

Figure 1.机理推测
在上述背景研究的基础上,Eiichi Nakamura、尚睿团队报道了第一例Fe催化等化学当量的(杂)芳烃和(杂)芳烃或带N-(喹啉-8-基)酰胺基团的烯烃在70℃条件下发生交叉偶联反应,能以高收率得到产物且不含任何自身偶联副产物。相关研究成果发表于。
Homocoupling-free iron-catalysed twofold C–H activation/cross-couplings of aromatics via transient connection of reactants
Doba,T.;Matsubara, T.; Ilies, L.; Shang, R.;* Nakamura, E.*Nature Catal. 2019, early view
DOI: 10.1038/s41929-019-0245-3.
论文作者介绍

研究者:Eiichi Nakamura教授
研究者经历
- 1973 B.S. Faculty of Science, Tokyo Institute of Technology (Professor TeruakiMukaiyama)
- 1978 Ph.D in chemistry, Department of Chemistry, Tokyo Institute of Technology (Professor Isao Kuwajima)
- 1978-1980 Postdoctoral Research Associate, Department of Chemistry, Columbia University, New York (Professor Gilbert Stork)
- 1980-1984 Assistant Professor, Department of Chemistry, Tokyo Institute of Technology
- 1984-1993 Associate Professor, Department of Chemistry, Tokyo Institute of Technology
- 1989-1991 Adjunct Associate Professor, Department of Applied Molecular Science, Institute for Molecular Science
- 1993-1995 Professor, Department of Chemistry, Tokyo Institute of Technology
- 1995-2016 Professor, Department of Chemistry, The University of Tokyo 2016-present Molecular Technology Innovation Chair Professor, The University of Tokyo
- 2004-2010 ERATO Nakamura Functional Carbon Cluster program research director, Japan Science and Technology Agency (JST)
- 2007-2012 The University of Tokyo, Chemistry Innovation Gobal COE Program Leader
- 2008-2009 Chairman, Department of Chemistry, The University of Tokyo
研究领域
- Physical Organic Chemistry/Synthetic Chemistry/Nano-science
- Major Research Interest Physical organic chemistry directed toward creation of new reactions, new molecules and materials, and new functions. Exploration of methodologies for physical organic chemistry such as high-resolution electron microscopy.
论文概要
以苯并[b]噻吩和3-甲基-N-(喹啉-8-基)苯甲酰胺为模板底物,作者对反应条件进行反复筛选,确定最佳条件为(Table 1):20 mol% Fe(acac)3和共轭双膦配体dppen作为最优催化剂,2.2 equiv 有机锌试剂Zn(CH2SiMe3)2⋅2MgCl2和1 equiv Me3SiCH2MgCl为最优碱,2 equiv DCP作为温和的氧化剂,THF为最优溶剂,在70 ℃条件下反应18 h,能以97%的收率得到目标产物。

Table 1铁催化的双重C-H活化/交叉偶联及其在材料合成中的应用。
各种C-2、C-3位苯基取代的噻吩、各种苯并[b]噻吩、苯并呋喃以及带给电子的芳烃甲酰胺均能很好地适应反应条件,可以良好的收率得到相应单芳基化产物。吲哚-2-甲酰胺、噻吩-2-甲酰胺与噻吩衍生物发生偶联反应,能以较好的收率得到双杂芳烃化合物。环烯酰胺以及非环状链烯醇也能较好的适应反应条件,能以良好的收率得到Z-构型产物。1-甲基-1H-吡唑则只能以较低的收率得到相应产物。苯和卤代苯衍生物也能适应反应条件,但只能以较低的收率得到相应产物。考虑到瞬态连接的有效性,作者应用该策略能快速得到高收率的多噻吩化合物,且通过简单的步骤,这些化合物可被转化为有用的空穴传输材料、导电聚合体等结构。
紧接着,作者做了一系列氘代实验。通过该实验数据,作者推测可能的机理:酰胺阴离子A可转化为氮杂金属环B,完成第一次C-H键的活化。中间体B和C可以快速达到平衡,且该过程可逆。紧接着,C会发生构型改变转化为C′,然后–CH2SiMe3基团可促使酰胺C-H键发生不可逆的去质子化反应,形成中间体D。最后D发生交叉偶联反应得到目标产物(Figure 2)。


Figure 2. 氘代实验及反应机理解析
论文总结评价
Eiichi Nakamura、尚睿团队报道了第一例Fe催化等化学当量的(杂)芳烃和(杂)芳烃或带N-(喹啉-8-基)酰胺基团的烯烃发生交叉偶联反应,能以高收率得到产物且不含任何自身偶联副产物。该反应为完全抑制自身偶联副产物的生成提供了一种强有力的策略,且通过多重C-H键激活/ C-C键交叉偶联反应得到的噻吩衍生物可转化为有用的空穴传输材料、导电聚合体等结构。
参考文献
- Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787. DOI: 10.1021/acs.chemrev.6b00567
- Shang, R.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2016, 138, 10132. DOI:10.1021/jacs.6b06908
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
N-磺酰基-1,2,3-三唑在羧酸铑催化剂作用下、能够产生含有亲核性的imine基团的卡宾中间体。该imina-carbenoid中间体可以进而发生跨环或者1,3-插入反应。
基本文献
Review
- Davies, H. M. L.; Alford, J. S. Chem. Soc. Rev. 2014, 43, 5151. DOI:10.1039/C4CS00072B
- Chattopadhyay, B.; Gevorgyan, V. Angew. Chem., Int. Ed. 2012, 51, 862. DOI:10.1002/anie.201104807
背景: 从三唑转化为卡宾活性中间体
1,2,3-三唑可以通过叠氮与炔烃的click反应进行合成、是一种芳香族化合物[3]。

芳虽然芳香族化合物具有固有的稳定性,但在N-原子上具有吸电子基团的三唑通过开环-闭环平衡反应得到微量的开环重氮亚胺[4]。与一般的重氮化合物类似,微量的开环重氮亚胺在金属催化剂的作用下可以产生金属卡宾化合物。也就是说碳原子对向金属提供 σ 孤对电子、然后再从金属方提供 π 电子,进而以离去氮气的形式形成下图所示的Imine-carbenoid活性中间体[5]。这种由N-取代三唑形成的卡宾中间体由于含有亚胺基团,因此被称为亚胺-卡宾中间体。

反应机理: “捕捉卡宾”→亚胺氮亲核进攻
亚胺-卡宾是一个含有强亲电子的卡宾部位与亲核性的亚胺氮部位的活性中间体。由于其氮原子参与反应,可以发生跨环与1,3-插入反应[6]。如下图所示,首先金属卡宾部位被富电子化合物或基团捕捉。然后 C—M σ 结合电子导向亚胺基团,使得亚胺的氮原子发生亲核进攻。
如果底物 X—Y 间具有多重结合键,那么发生跨环反应形成含氮杂环化合物。如果X—Y 间是单键那么发生1,3-插入反应得到链状胺产物。

反应实例
跨环反应

关于含氮杂环化合物的合成有许多报道[2]。具体的有以末端烯烃[7]、富电子烯烃[8, 9]、醛[10]、氰基[11]等为底物分别合成吡咯,二氢吡咯,恶唑啉,咪唑。
1,3-插入反应

单纯的例子有,对底物醇,羧酸,一级酰胺进行的 X—H 键插入[12]与烯丙基或苄基化合物的C—H 插入[13]。然而,在许多情况下,这些1,3-插入产物随后会经历互变异构或重排反应。例如在与水发生1,3-插入生成的烯胺发生互变异构最终得到氨基酮(上图左端, R3=H)[14]。另外,当与羧酸反应时,羧酸底物的取代基 R3 立体位阻较小的情况,会发生分子内缩合反应,酰基会重排到N上 (上图从左开始第二个反应, 反应机理参考下图)[12]。

变体:叠氮化物和炔烃的一锅反应
叠氮化物和炔烃的点击反应后,不后处理直接加入催化剂和另一种反应底物、利用该one-pot反应可以用来合成氨基酮[14]。

实验技巧
三唑的合成原料叠氮具有爆炸性,取用时候要注意。
三唑由于结晶性比较高,虽然可以在冷暗处保存,但是容易在空气中发生微量的水解,因此在grove-box里面操作更佳。另外其水解物容易对金属卡宾的生成起阻碍作用,在使用前最好进行重结晶纯化。
关联反应
- Huisgen Cycloaddition
- Dimroth Rearrangement via A Conjugated 1,3-Dipole
- C—H Insertion of Metal Carbenoid
参考文献
- Davies, H. M. L.; Alford, J. S. Chem. Soc. Rev. 2014, 43, 5151. DOI:10.1039/C4CS00072B
- Chattopadhyay, B.; Gevorgyan, V. Angew. Chem., Int. Ed. 2012, 51, 862. DOI:10.1002/anie.201104807
- Raushel, J.; Fokin, V. V. Org. Lett. 2010, 12, 4952. DOI: 10.1021/ol102087r
- [a] Harmon, R. E.; Stanley, F. Jr.; Gupta, S. K.; Johnson, J. J. Org. Chem. 1970, 35, 3444. DOI: 10.1021/jo00835a057 [b] L’abbé, G. Bull. Soc. Chim. Belg. 1990, 99, 281–290. DOI: 10.1002/bscb.19900990410
- Nakamura, E.; Yoshikai, N.; Yamanaka, M. J. Am. Chem. Soc. 2002, 124, 7181.DOI: 10.1021/ja017823o
- 跨环是Transannulation的中译
- Chattopadhyay, B.; Gevorgyan, V. Org. Lett. 2011,13, 3746. DOI: 10.1021/ol2014347
- Kwok, S. W.; Zhang, L.; Grimster, N. P.; Fokin, V. V. Angew. Chem., Int. Ed. 2014, 53, 3452. DOI: 10.1002/anie.201306706
- 从烯烃得到二氢吡咯的反应,详细参考文献[8].

- Zibinsky, M.; Fokin, V. V. Angew. Chem., Int. Ed. 2013, 52, 1507. DOI: 10.1002/anie.201206388
- Horneff, T.; Chuprakov, S.; Chernyak, N.; Gevorgyan, V.; Fokin, V. V. J. Am. Chem. Soc. 2008, 130, 14972. DOI; 10.1021/ja805079v
- Chuprakov, S.; Worrell, B. T.; Selander, N.; Sit, R. K.; Fokin, V. V. J. Am. Chem. Soc. 2014, 136, 195. DOI: 10.1021/ja408185c
- Kubiak, R. W. II; Mighion, J. D.; Wilkerson-Hill, S. M.; Alford, J. S.; Yoshidomi, T.; Davies, H. M. L. Org. Lett. 2016, 18, 3118. DOI: 10.1021/acs.orglett.6b01298
- Miura, T.; Biyajima, T.; Fujii, T.; Murakami, M. J. Am. Chem. Soc. 2012, 134, 194. DOI: 10.1021/ja308285r
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
由芳基或烯基卤化物/三氟甲磺酸酯作为底物与钯催化剂合成有机硼酸化合物的方法。市售的频哪醇二硼化合物(PinB-BPin)常常作为硼源。另一方面使用H-BPin、(HO)2B-B(OH)2作为硼源的条件也有所报道。
近年来,随着铱催化剂的开发应用,不通过卤素,直接经由C-H活化进行硼酸化的反应被开发(Hartwig·Miyaura硼化)。 请参阅关联反应。
关联反应
- Liebeskind-Srogl Cross Coupling
- Cross Dehydrogenative Coupling (CDC)
- Catalytic C-H Oxidation
- Hofmann-Löffler-Freytag Reaction
- Catalytic C-H activation
- C-H Insertion of Metal Carbenoid
- Brown Hydroboration
- Suzuki-Miyaura Cross Coupling
- Hartwig-Miyaura C-H Borylation
基本文献
- Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508. DOI: 10.1021/jo00128a024
- Murata, M.; Watanabe, S.; Masuda, Y. J. Org. Chem. 1997, 62, 6458. DOI: 10.1021/jo970963p
- Murata, M.; Oyama, T.; Watanabe, S.; Masuda, Y. J. Org. Chem. 2000, 65, 164. DOI: 10.1021/jo991337q
- Takagi, J.; Takahashi, K.; Ishiyama, T.; Miyaura, N. J. Am. Chem. Soc. 2002, 124, 8001. DOI: 10.1021/ja0202255
<review>
- Ishiyama, T.; Miyaura, N. Chem. Rec. 2004, 3, 271. DOI: 10.1002/tcr.10068
- Shimizu, M.; Hiyama, T. Eur. J. Org. Chem. 2013, 36, 8096. DOI: 10.1002/ejoc.201300632
- Chow, W. K.; Yuen, O. Y.; Choy, P. Y.; So, C. M.; Lau, C. P.; Wong, W. T.; Kwong, F. Y. RSC Adv. 2013, 3, 12518. DOI: 10.1039/C3RA22905J
- Shinokubo, H. Proc. Jpn. Acad. Ser. B. 2014, 90, 1. doi:10.2183/pjab.90.1
反应机理
基本反应机理与钯交叉偶联相似。具体请参照Suzuki-Miyaura Cross Coupling。

反应实例
通过选择具有适当反应性的底物,可以经过Miyaura硼酸化→Suzuki-Miyaura交叉偶联的连续反应获得联芳烃。

适用于低反应性氯代芳烃的反应条件[1]

实验步骤
芳基溴化物的硼化[2]

实验技巧
※碱的话KOAc是最优的。在使用像K2CO3,K3PO4这样相对强的碱的话,会伴随形成铃木偶联形式的二聚体副产物。对三氟甲磺酸烯基酯作为底物的时候,使用KOPh做为碱进行反应。
※对于溶剂,反应速度如下 DMSO >> DMF > 1,4-dioxane。
参考文献
- (a) Billingsley, K. L.; Barder, T. E.; Buchwald, S. L. Angew. Chem. Int. Ed. 2007, 46, 5359. doi:10.1002/anie.200701551 (b) Billingsley, K. L.; Buchwald, S. L. J. Org. Chem. 2008, 73, 5589. DOI: 10.1021/jo800727s
- Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508. DOI: 10.1021/jo00128a024
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!