概要
Marshall 炔丙基化是炔丙基甲磺酸酯还原条件下与醛基发生偶联,产生碳-碳键的反应。甲磺酸酯所在的碳原子的手性翻转,生成的产物有两个不对称中心。反应常用双乙基锌、InI(I)等作为还原剂,并且可以大量合成。
基本文献
- Marshall, J. A.; Adams, N. D. J. Org. Chem. 1998, 63, 3812. DOI: 10.1021/jo980623j
- Marshall, J. A.; Adams, N. D. J. Org. Chem. 1999, 64, 5201. DOI: 10.1021/jo9823083
- Marshall, J. A.; Grant, C. M. J. Org. Chem. 1999, 64, 696. DOI: 10.1021/jo982255p
- Marshall, J. A.; Chobanian, H. R.; Yanik, M. M. Org. Lett. 2001, 3, 3369. DOI: 10.1021/ol016605x
- Marshall, J. A.; Schaaf, G. M. J. Org. Chem. 2001, 66, 7825. DOI: 10.1021/jo015936k
<review>
- Marshall, J. A. J. Org. Chem. 2007, 72, 8153. DOI: 10.1021/jo070787c
- Ding, C.-H.; Hou, X.-L. Chem. Rev. 2011, 111, 1914. DOI: 10.1021/cr100284m
反应机理
通过钯催化剂氧化还原过程能够生成立体特异性的手性丙二烯金属中间体,再与醛通过环状过渡中间体发生手性翻转,并生成新的碳-碳键。

反应实例
Iejimalide B的合成[1]

Ushikulide A的全合成[2]

参考文献
- Chen, Q.; Schweitzer, D.; Kane, J.; Davisson, V. J.; Helquist,P. J. Org. Chem. 2011, 76, 5157. DOI: 10.1021/jo200514m
- (a) Trost, B. M.; O’Boyle, B. M. J. Am. Chem. Soc. 2008, 130, 16190. (b) Trost, B. M.; O’Boyle, B. M.; Hund, D. J. Am. Chem. Soc. 2009, 131, 15061. DOI: 10.1021/ja906056v
外部链接
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
概要
利用铱金属催化剂与给电子性双齿配体,可以不经过芳香卤代烃这一中间体直接进行C-H键活化硼基化反应。作为生成物的有机硼化合物可以作为多种化学官能团转变的中间体,有非常高的化学利用价值。
本反应容易受到芳香环上取代基的立体位阻的影响。因此,ortho-金属化及亲电取代反应较难发生,结果上来看,它可以实现取代芳基的间位选择性官能团化。
利用芳香卤代物做中间体的反应见Miyaura-Ishiyama硼基化反应。
基本文献
- Cho, J. Y.; Tse, M. K.; Holmes, D.; Maleczka, R. E., Jr.; Smith, M. R. Science 2002, 295, 305. DOI:10.1126/science.1067074
- Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc.2002, 124, 390. DOI: 10.1021/ja0173019
- Ishiyama, T.; Takagi, J.; Hartwig, J. F.; Miyaura, N. Angew. Chem. Int. Ed. 2002, 41, 3056. [abstract]
- Ishiyama, T.; Nobuta, Y.; Hartwig, J. F.; Miyaura, N. Chem. Commun. 2003, 2924. DOI: 10.1039/B311103B
<review>
- Ishiyama, T.; Miyaura, N. Chem. Rec. 2004, 3, 271. DOI: 10.1002/tcr.10068
- Ishiyama, T.; Takagi, J.; Nobuta, Y.; Miyaura, N. Org. Synth. 2005, 82, 126. DOI: 10.15227/orgsyn.082.0126
- Mkhalid, I. A. I.; Barnard, J. H.; Marder, T. B.; Murphy, J. M.; Hartwig, J. F. Chem. Rev. 2010, 110,890. doi:10.1021/cr900206p
- Hartwig, J. F. Chem. Soc. Rev. 2011, 40, 1992. doi:10.1039/C0CS00156B
- Hartwig, J. F. Acc. Chem. Res. 2012, 45, 864. DOI: 10.1021/ar200206a
- Ros, A.; Femandez, R.; Lassaletta, J. M. Chem. Soc. Rev. 2014, 43, 3229. DOI: 10.1039/C3CS60418G
- Shinokubo, H. Proc. Jpn. Acad. Ser. B. 2014, 90, 1. doi:10.2183/pjab.90.1
- Yang, J. Org. Biomol. Chem. 2015, 13, 1930. DOI: 10.1039/C4OB02171A
反应机理
铱・配体・Bpin-Bpin(双联频哪醇硼酸酯)混合后生成的fac-trisboryl iridium complex中间体是能催化反应的活性物种。烯烃配体(COD等)离去后会给六配位的Ir(III)留一个空缺位,使得C-H键在此通过氧化加成反应活化。(参考:J. Am. Chem. Soc. 1993, 115, 9329; J. Am. Chem. Soc. 2003, 125, 16114; J. Am. Chem. Soc. 2005, 127, 14263 )

反应实例
因为主要受立体因素的影响,一般在取代基的邻位硼基化反应较为困难。(图片来自这里)

卟啉的β位选择性C-H官能团化[1]


氢硅烷基作为指向基,实现通常条件下较难发生的邻位C-H键硼基化反应。[3]

指向性官能团的存在下,环丙烷[4]以及其他sp3C-H键的硼基化反应也能进行。[5]

通过对催化剂的调整、以醇羟基做指向基团可活化不太容易活化的sp3C-H键的硅基化[6],之后再通过玉尾氧化转变为醇、这一转化作为一种新型多醇合成法有着很高的利用价值,以下是其在糖合成反应中的应用实例。[7]

使用大位阻的双齿膦配体与金属铱催化剂能够实现para位的C-H键硼基化反应[8]

用铑催化剂可实现烷烃的终端惰性C-H键的官能团化。[9]

参考文献
- [1] Hata, H.; Shinokubo, H.; Osuka, A. J. Am. Chem. Soc. 2005, 127, 8264. DOI: 10.1021/ja051073r
- [2] Fischer, D.F.; Sarpong, R. J. Am. Chem. Soc. 2010, 132, 5926. DOI: 10.1021/ja101893b
- [3] Boebel, T. A.; Hartwig, J. F. J. Am. Chem. Soc. 2008, 130, 7534.DOI:10.1021/ja8015878
- [4] Liskey, C. W.; Hartwig, J. F. J. Am. Chem. Soc. 2013, 135, 3375. DOI: 10.1021/ja400103p
- [5] (a) Liskey, C. W.; Hartwig, J. F. J. Am. Chem. Soc. 2012, 134, 12422. DOI: 10.1021/ja305596v (b) Li, W.; Liskey, C. W.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 8755. DOI: 10.1021/ja503676d (c) Miyamura, S.; Araki, M.; Suzuki, T.; Yamaguchi, J.; Itami, K. Angew. Chem. Int. Ed. 2015, 54, 846. DOI: 10.1002/anie.201409186
- [6] (a) Simmon, E. M.; Hartwig, J. F. Nature 2012, 483, 70. doi:10.1038/nature10785 (b) Li, B.; Driess, M.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 6586. DOI: 10.1021/ja5026479
- [7] Frihed, T. G.; Heuckendorff, M.; Pedersen, C. M.; Bols, M. Angew. Chem. Int. Ed. 2012, 51, 12285. DOI: 10.1002/anie.201206880
- [8] Saito, Y.; Segawa, Y.; Itami, K. J. Am. Chem. Soc. 2015, 137, 5193. DOI: 10.1021/jacs.5b02052
- [9] Chen, H.; Schlecht, S.; Semple, T. C.; Hartwig, J. F. Science 2000, 287, 1995. DOI:10.1126/science.287.5460.1995
关联书籍
[amazonjs asin=”0471112801″ locale=”US” title=”Organic Synthesis Via Boranes”] [amazonjs asin=”3527325980″ locale=”US” title=”Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials”] [amazonjs asin=”3527331549″ locale=”US” title=”Metal Catalyzed Cross-Coupling Reactions and More, 3 Volume Set”] [amazonjs asin=”148223310X” locale=”US” title=”C-H Bond Activation in Organic Synthesis”]
相关反应
- 金属催化C-H活化反应 Catalytic C-H activation
- Brown硼氢化-氧化反应 Brown Hydroboration
- 铃木-宮浦偶联反应 Suzuki-Miyaura Cross Coupling
- 宫浦硼化反应 Miyaura-Ishiyama Borylation
外部链接
- Ir-catalyzed C-H borylation (Sigma-Aldrich)
- John F. Hartwig Group
- Borylation – Wikipedia
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
概要
过渡金属催化的导向碳氢键官能化反应由于其步骤经济性以及碳氢键在有机分子中的普遍存在的特性,展示出了广阔的应用前景。导向的直接碳氢键官能化主要通过环金属化的金属有机中间体进行,虽然环金属化反应多报道于后过渡金属,但是含有Csp2-铁和Csp3-铁键的铁碳环金属有机化合物也有过报道。E. Nakamura等人首次发现通过生成铁碳环中间体可以实现杂原子导向的C-H活化C-C键生成反应[1, 2]。铁作为地壳中储量最大的过渡金属元素,由于其大储量以及廉价无毒的特性,铁催化作为一种环境友好且可持续的催化剂[3, 4],在碳氢键活化中的运用近年开始受到了广泛的关注。目前该反应已经拓展到Csp2-H以及Csp3-H与亲核试剂以及亲电试剂[6, 7]的反应,展示出了潜在的发展潜力和应用前景。

基本文献
- [1] Norinder, J.; Matsumoto, A.; Yoshikai, N.; Nakamura, E. J. Am. Chem. Soc. 2008, 130, 5858. DOI: 10.1021/ja800818b
- [2] Yoshikai, N.; Matsumoto, A.; Norinder, J.; Nakamura, E. Angew. Chem., Int. Ed. 2009, 48, 2925. DOI: 10.1002/anie.200900454
- [3] Nakamura, E.; Yoshikai, N. J. Org. Chem. 2010, 75, 6061. DOI: 10.1021/jo100693m
反应机理
- 该类型的铁催化导向碳氢官能化反应与以往的很多自由基类型的铁催化反应不同,该类铁催化的碳氢键活化是通过与贵金属Pd,Rh等类似的环金属化(cyclometallation)中间体进行。反应涉及到铁碳环的生成,以及铁碳环参与的还原消除或者氧化加成反应。由于铁金属有机中间体在分离上的困难,目前该反应的机理并不是十分明确。研究指出针对具体的情况“还原的低价铁”以及三价铁都可以作为活化碳氢键的活性物种。反应中活化碳氢键所使用的碱目前主要依赖于金属有机试剂这一类强碱。1,2-二氯代烷烃常作为该反应中氧化铁催化剂的氧化剂。

反应实例
- 铁催化的氮原子导向的直接碳氢键芳基化反应[1]

- 铁催化的非自由基C(sp3)-H直接芳基化[5]

- 铁催化的直接碳氢活化烯丙基化(与亲电试剂直接偶联)[7]

实验步骤
以反应实例1为例。在零度的冰水浴中,在惰气保护下,往0毫摩尔的反应物的无水无氧四氢呋喃溶液中加入乙酰丙酮铁以及配体,得到反应物和催化剂的混合物。在另一个Schlenk反应管中将格式试剂与锌盐混合并搅拌20分钟。将预先混合的催化剂和底物的混合溶液转移到制备的锌试剂溶液中。最后往该反应混合物中加入1,2-二氯异丁烷,然后在冰水浴中搅拌16小时。反应结束后,通过加入酒石酸钠钾的饱和水溶液淬灭反应。通过柱层析分离得到反应产物。
实验技巧
由于该类反应使用到金属有机试剂,如格氏试剂和锌试剂,反应对于溶剂的干燥程度要求比较高。使用新制的金属有机试剂是更佳的选择,这是由于久置的金属有机试剂往往由于保管不当而含有金属氢氧化物,这会抑制铁催化剂的活性。
参考文献
- [4] Ilies, L.; Nakamura, E. Iron-Catalyzed Cross-Coupling Reactions, in The Chemistry of Organoiron Compounds; Eds. Marek, I.; Rappoport, Z.; John Wiley & Sons, Ltd.: Chichester, UK, 2014.
- [5] Shang, R.; Ilies, L.; Matsumoto, A.; Nakamura, E. J. Am. Chem. Soc. 2013, 135, 6030. DOI: 10.1021/ja402806f
- [6] Matsubara, T.; Asako, S.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2014, 136, 646. DOI: 10.1021/ja412521k
- [7] Asako, S.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2013, 135, 17755. DOI: 10.1021/ja4106368
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
对惰性的C-H键进行直接的切断,重组,连接上不同的官能团的十分有用的手法。
由于是直接C-H活化,所以底物不需要进行卤代,而且副产物也随着方法的改善而变得越来越可控。此类反应绝对将在未来有机合成中扮演最重要的角色之一。
现今虽然这方面研究是一大热点,经常有很多文章出现,但是总体来说反应条件还是比较苛刻,为了实现化学选择性经常需要引入特定的导向基团,所以大范围使用还是受到很大的限制。而今后的研究也主要着重于解决这一类问题。
基本文献
- Murai, S.; Kakiuchi, F.; Sekine, S.; Tanaka, Y.; Kamatani, A.; Sonoda, M.; Chatani, N. Nature 1993,366, 529. doi:10.1038/366529a0
- Campeau, L. -C.; Stuart, D. R.; Fagnou, K. Aldrichimica Acta 2007, 40, 35. [PDF]
- Alberico, D.; Scott, M. E.; Lautens, M. Chem. Rev. 2007, 107, 174. DOI: 10.1021/cr0509760
反应机理
在近位有导向基团存在的条件下,实现金属配位促进氧化加成并且得到选择性产物。
反应实例
近年,没有卤代或者硼酸化修饰的底物的直接的coupling的研究十分火热。Fagnou等人,开发出了donar・acceptor无修饰型底物的直接C-H活化偶联反应[1]。

sp3-sp3碳-碳偶联的杂原子胺基烷基化反应。[2]

实验步骤
α-萘酮的邻位C-H活化[3]

实验技巧
参考文献
- (a) Fagnou, K. et al. Science 2007, 316, 1172. DOI: 10.1126/science.1141956 (b) Fagnou, K. et al. J. Am. Chem. Soc. 2007, 129, 12072. DOI: 10.1021/ja0745862
- Herzon, S. B.; Hartwig, J. F. J. Am. Chem. Soc.2007, 129, 6690. DOI: 10.1021/ja0718366
- Kakiuchi, F.; Murai, S. Org. Synth. 2002, 80, 104. [PDF]
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
- 概要
烯/炔作为反应底物的时候,仅仅从合成化学的观点来看,M可以广泛的是 B(Brown加氢硼化)、Al(加氢铝化)、Zr(加氢锆化)、Sn(加氢锡化)、Si(加氢硅化)。通常反应按照顺势加成形式进行,有些特别的情况也观测到了反式加成的产物。Schwartz试剂(Cp2ZrHCl)是一种非常有名的加氢金属化试剂。
生成的烷基/烯基金属化合物自身是作为反应活性中间体,和亲电试剂反应能够变换出各种产物。
- 基本文献
- 反应机理

- 反应实例

加氢镁化一般是用二茂钛催化剂以及含有β氢的Grignard试剂实现。[1]它能用于合成一些用通常方法合成不了的Grignard试剂。

加氢锡化要用到金属催化剂反应才会进行。现在最常用到的是钯。它能用于立体选择性合成烯基碘化物。

- 实验步骤
- 实验技巧
- 参考文献
[2] Smith, A. B., III et al. J. Am. Chem. Soc. 2001, 123, 10942. DOI: 10.1021/ja011604l
- 概要
第一主族金属存在下,两分子的卤代烷烃发生还原自偶联生成新的烷烃的反应。还原剂除了第一主族的金属之外,Mg、Ni(CO)4、t-BuLi等也可以被使用。
本反应主要的副反应是消去反应。分子间反应发生的条件是一级卤化物,尤其是碘化物产率最好,第二级第三级卤化物的产率非常低。
芳基卤化物和烷基卤化物的组合会发生偶联反应(Wurtz-Fittig反応)。化学选择性取决于被还原的容易程度(芳基卤化物更容易被还原)。
![]()
- 基本文献
・Wurtz, A. Ann. Chim. Phys. 1855, 44, 275
・Wurtz, A. Ann. 1855, 96, 364. doi:10.1002/jlac.18550960310
・Tollens, B.; Fittig, R. Ann. Chem. Pharm. 1864, 131, 303. doi:10.1002/jlac.18641310307
・Hilingtion, D. C. Comprehensive Organic Synthesis 1991, 3, 413.
- 反应机理
一分子的烷基卤化物和碱金属生成有机金属试剂,然后与另一分子的烷基卤化物发生置换反应得到烷烃和盐。

- 反应实例

- 实验步骤
- 实验技巧
- 参考文献
烯烃→醛
- 概要
用一氧化碳和氢气的混合气体使烯基反应成为增长一个碳的醛的反应。常用的催化剂有铑或钴配体。钴催化剂相对比较便宜(Co2(CO)8),而铑比较贵但是催化活性更好,适合实验室小量反应研究使用。反应用到气体所以很适合工业生产,是非常实用的一个反应。
- 基本文献
・Evans, D.; Osborn, J. A.; Wilkinson, G. J. Chem. Soc. 1968, 33, 3133. doi:10.1039/J19680003133
・Ojima, I.; Tsai, C. Y.; Tzamarioudaki, D.; Bonafoux, D. Org. React. 2000, 56, 1.
- 反应机理

- 反应实例

不对称的磷或氧磷配体,尤其是Rh-BINAPHOS,能得到高对映选择性的带支链的醛。

(+)-Ambruticin 的合成[1]

- 实验步骤
- 实验技巧
往往用夹角较大的双齿磷配位的铑能够改善使反应趋向于生成支链的醛。
- 参考文献
- 概要
Tebbe试剂,是一种将羰基变换为双键的有用的试剂。 醛和酮能够被反应以外,用维蒂希试剂不能够反应的酯基,内酯,酰胺,硫酯的碳氧双键也能够被反应得到烯基。 Tebbe试剂碱性弱,路易斯酸性强。因此对于一些立体阻碍大的羰基,烯醇重排的羰基,α位碳具有手性的羰基,他都能够尽量避免差向异构化的发生而进行反应。 如果在恰当的位置有双键存在的话,会发生烯烃复分解反应。
- 基本文献
・ Tebbe, F. N.; Parshall, G. W.; Reddy, G. S. J. Am. Chem. Soc. 1978, 100, 3611. doi:10.1021/ja00479a061
・ Pine, S. H.; Zahler, R.; Evans, D. A.; Grubbs, R. H. J. Am. Chem. Soc. 1980, 102, 3270. DOI: 10.1021/ja00529a076
・ Brown-Wensley, K. A.; Buchwald, S. L.; Cannizzo, L.; Clawson, L.; Ho, S.; Meinhardt, D.; Stille, J. R.; Straus, D.; Grubbs, R. H. Pure Appl. Chem. 1983, 55, 1733. doi:10.1351/pac198355111733
・ Pine, S. H. et al. J. Org. Chem. 1985, 50, 1212. DOI: 10.1021/jo00208a013
・ Pine, S. H. et al. Org. Synth. 1990, 65, 72. ・ Kelly, S. E. Comp. Org. Syn. 1991, 1, 743.
・ Pine, S. H.; Shen, G. S.; Hoang, H. Synthesis 1991, 165. DOI: 10.1055/s-1991-26406 ・ Pine, S. H. Org. React. 1993, 43, 1.
・ Beadham, I.; Micklefield, J. Curr. Org. Syn. 2005, 2, 231.
- 反应机理
反应活性体是Cp2Ti=CH2。它和羰基反应,生成双键。这是由于金属和氧的强成键作用促进了反应的发生。
- 反应实例

- 实验步骤
Tebbe试剂合成方法:二氯二环戊二烯钛(Cp2TiCl2)和三甲基铝在甲苯中混合制备。 ※该试剂有引火性,在惰性气体下使用。
※该试剂有引火性,在惰性气体下使用。
- 实验技巧
- 参考文献
- 概要
α-卤代酯和锌生成的锌的烯醇盐与醛酮的加成反应。
与烯醇锂、烯醇镁等相比,锌的烯醇盐碱性弱,活性较低,官能团兼容性高。对酯类化合物反应能进行但比较慢。
近年来,也有发现除锌以外的金属也能发生类似的反应,特别是Sm(II)、Cr(II)、Ti(II)等常使用。
- 基本文献
・ Reformatsky, S. Ber. 1887, 20, 1210. doi:10.1002/cber.188702001268
・ Reformatsky, S. J. Russ. Phys. Chem. Soc. 1890, 22, 44.
・ Review: Shriner, R. L. Org. React. 1942, 1, 1.
・ Review: Rathke, M. W. Org. React. 1975, 22, 423.
・ Review: Furstner, A. Synthesis 1989, 571. DOI: 10.1055/s-1989-27326
・ Review: Rathke, M. W. Comprehensive Organic Synthesis 1991, 2, 277.
・ Review: Ocampo, R.; Dolbier, W. R. Tetrahedron 2004, 60, 9325. doi:10.1016/j.tet.2004.07.018
- 反应机理
体系中生成的锌的烯醇盐,醚溶剂中是以二聚体形式存在,C-烯醇盐的存在形式已经由图谱,结晶构造解析来证实。下图是二聚体的解离,反应时一个单体与醛酮的氧经由六元环过渡态来完成的。

- 反应实例
用SmI2分子内的Reformatsky反应[1]

- 实验步骤
- 实验技巧
- 参考文献
- 概要
由一价铜和两当量的有机锂试剂制备的有机铜锂试剂(organocuprate)[R2CuLi],亲核性强,用于α,β-不饱和羰基化合物的1,4-加成反应、还有sp3碳上的取代反应,反应都能很快进行。
有机铜试剂碱性弱,脱氢化等副反应较难发生。而单用有机锂试剂1,2-加成优先发生,两者辅助使用,可以实现1,4-加成。
该试剂反应性非常高、对于立体位阻大的碳原子也可以反应。通过TMSCl等硬路易斯酸,可以加速1,4-加成反应。
除了与有机锂试剂以外,也可以与格利雅试剂、有机锌试剂等一起使用。特别是和后两者结合使用时,可以将铜试剂的量减小到催化剂量。
两当量的有机锂试剂对于反应的顺利进行很必要,实际仅有一当量在反应中消耗。通过导入较难迁移的配体(dummy ligand)生成杂有机铜试剂 (mixed organocuprate)[R(X)CuLi](X = alkenyl, -CN, -SR’,-NR’2, PR’2 etc.)、可以对重要的反应剂高效利用。
近年来,通过催化剂量的铜-手性膦配体的使用,发展了许多不对称1,4-加成反应。
- 基本文献
・Modern Organocopper Chemistry, Krause, N. Ed.; Wiley-VCH; 2002.
・Posner, G. H. Org. React. 1972, 19, 1.
・Posner, G. H. Org. React. 1975, 22, 253.
- 反应机理
有机铜试剂的结构根据溶剂体系的不同,会有些差异。速度论实验等得结果表明、二聚体[R2CuLi]2很有可能是反应中的可能活性中间体
近几年、中村等、报道了由计算化学研究的详细反应机理(参考: Angew. Chem. Int. Ed. 2000, 39, 3750. 有機合成化学協会誌, 2003, 61, 144.)。
1,4-加成反应中、生成dCu-π*C=C络合物形成后,富电子的一价铜Cu(I)发生氧化加成、生成Cu(III)中間体。近年、Cu(III)中间体的结构研究由分光分析及理论计算的推测(参考:J. Am. Chem. Soc. 2007,129, 7208; J. Am. Chem. Soc. 2007, 129, 7210.)。 接下来的还原消去得到金属烯醇盐,这一过程是该反应的决速步骤。

「dummy ligand 因为和铜较强的作用不发生转移。」这一解释能够行的通。近年、锂和正离子的π相互作用使dummy ligand向不发生迁移的方向固定。这样全新的解释是中村等通过计算提出的。

置换反应中,经由铜(III)中间体的反应机理,通过实验,计算两方面得到证实。

- 反应实例
环状不饱和酮时, 受取代基立体位阻的影响、反应进行具有立体选择性。d-π*络合物的形成是高立体选择性的关键。


1,4-加成后生成的金属烯醇盐具有活性、然后加入亲电试剂后,发生一锅法三组分连结型反应。下图是用此法合成前列腺素的实例[1]。
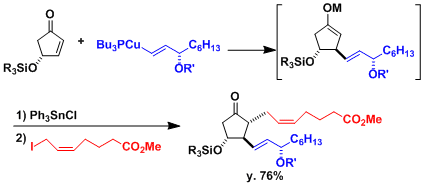
用CuCN作为铜源制备的[R2Cu(CN)Li2]、被称为higher order cuprate(Lipshutz cuprate)、与通常的铜试剂相比具有更高的反应性和化学选择性。
![]()
烯基卤代烃・OTf的sp2碳上发生取代(偶联)反应。
![]()
TMSCl或BF3等路易斯酸共存下、立体位阻较大的位置也能发生共轭加成。[2] 位置选择性的生成烯醇中间体。

- 实验步骤

环氧化物(3.50g, 40.6 mmol)的THF溶液(30mL)中、加入CuCN(364mg, 3.65mmol)。冷却至-78℃下,边搅拌边由滴液漏斗缓慢加入乙烯溴化镁(1M in THF, 52.8mL, 52.8 mmol),45分滴加完成。反应混合物升至0℃、加入饱和氯化铵水溶液(20mL)。分离有机相,水相用乙醚萃取三次,与有机相合并后用饱和食盐水洗涤,无水硫酸钠干燥。过滤后、减压浓缩,所得粗产物用柱色谱(乙醚/戊烷=1/3)纯化分离。蒸除溶剂后得到目标产物为淡黄色液体(4.41g, 収率95%)。[2]
- 实验技巧
有机铜试剂对热不稳定,升温加热的话会发生铜试剂烷基与烷基的偶联,迅速分解。该试剂不能保存,必须现用现制。
- 参考文献
[2] (a) Yamamoto, Y. Angew. Chem. Int. Ed. 1986, 25, 947. (b) Lipshutz, B. H.; Ellsworth, E. L.; Siahaan, T. J. Am. Chem. Soc. 1989, 111, 1351.
[3]Holub, N.; Neidhorfer, J.; Blechert, S. Org. Lett. 2005, 7, 1227.