作者:杉杉
导读:
近日,英国Bristol大学的V. K. Aggarwal课题组在Angew. Chem. Int. Ed.中发表论文,报道一种全新的通过张力释放驱动的采用BCB (bicyclo[1.1.0]butane)分子参与的环氧化与氮杂环丙烷化反应方法学,进而成功完成一系列螺环环氧化物与螺环氮杂环丙烷分子的构建。
Strain-Release Driven Epoxidation and Aziridination of Bicyclo[1.1.0]butanes via Palladium Catalyzed σ-Bond Nucleopalladation
B.Wölfl, N.Winter, J. Li, A. Noble, V. Aggarwal, Angew. Chem. Int. Ed. 2022, ASAP. doi: 10.1002/anie.202217064.

正文:
目前,通过螺环环氧化物参与的合成转化方法学研究,已经备受广泛关注 (Scheme 1A)[1]-[3]。这里,受到近年来对于通过环丁酮砌块构建1-氧杂螺[2.3]己烷分子[4]-[5]以及1,1,3-三取代环丁烷分子模块化合成 (modular synthesis)反应方法学[6]-[7] (Scheme 1B)相关研究报道的启发,英国Bristol大学的V. K. Aggarwal课题组成功设计出一种全新的通过张力释放驱动的采用BCB (bicyclo[1.1.0]butane)分子参与的环氧化与氮杂环丙烷化反应方法学 (Scheme 1C)。

首先,作者采用苯丙酮9与亚砜衍生物10作为模型底物,进行相关反应条件的优化筛选 (Scheme 2)。进而确定最佳的反应条件为:在-95 oC (液氮-丙酮浴)条件下,将叔丁基锂的戊烷溶液滴加至BCB-亚砜衍生物10的THF溶液中,并剧烈搅拌1 min,之后再滴加苯丙酮9的THF溶液,接下来,再次向冷浴中加入干冰,并继续搅拌1 h。移开冷浴后,再加入Ph-OTf,之后加入含有Pd(dba)2与dippf的THF溶液,并升温至60 oC,反应18 h后,最终获得相应的1-氧杂螺[2.3]己烷衍生物18。

之后,基于前期相关的文献报道[8],作者提出如下合理的反应机理 (Scheme 3)。

在上述的最佳反应条件下,作者分别对一系列酮底物以及三氟甲磺酸酯底物的应用范围进行深入研究 (Scheme 4)。

同时,作者对一系列醛与亚胺底物 (Scheme 5)的应用范围进行深入研究。

之后,该小组通过如下的一系列研究进一步表明,这一全新的环氧化与氮杂环丙烷化策略具有潜在的合成应用价值 (Scheme 6)。

总结:
英国Bristol大学的V. K. Aggarwal团队成功设计出一种全新的通过张力释放驱动的采用BCB (bicyclo[1.1.0]butane)分子参与的环氧化与氮杂环丙烷化反应方法学,进而成功完成一系列螺环环氧化物与螺环氮杂环丙烷分子的构建。这一全新的环氧化与氮杂环丙烷化具有广泛的底物应用范围以及良好的合成应用价值等优势。
参考文献:
- [1] E. Leemans, M. D′hooghe, N. De Kimpe, Chem. Rev. 2011, 111, 3268. doi: 10.1021/cr100295j.
- [2] Z. Li, V. Gevorgyan, Angew. Chem. Int. Ed. 2012, 51, 1225. doi: 10.1002/anie.201106969.
- [3] S. Ahmad, W. N. Washburn, A. S. Hernandez, S. Bisaha, K. Ngu, W. Wang, M. A. Pelleymounter, D. Longhi, N. Flynn, A. V. Azzara, K. Rohrbach, J. Devenny, S. Rooney, M. Thomas, S. Glick, H. Godonis, S. Harvey, H. Zhang, B. Gemzik, E. B. Janovitz, C. Huang, L. Zhang, J. A. Robl, B. J. Murphy, J. Med. Chem. 2016, 59, 8848. doi: 10.1021/acs.jmedchem.6b00676.
- [4] A. Cocco, M. G. Rubanu, M. L. Sechi, A. Frongia, P. Mastrorilli, L. Degennaro, M. Colella, R. Luisi, F. Secci, Org. Biomol. Chem. 2021, 19, 1945. doi: 10.1039/D0OB00771D.
- [5] Y. Liu, H. Li, S. Chiba, Org. Lett. 2021, 23, 427. doi: 10.1021/acs.orglett.0c03935.
- [6] L. Lewis-Borrell, M. Sneha, I. P. Clark, V. Fasano, A. Noble, V. K. Aggarwal, A. J. Orr-Ewing, J. Am. Chem. Soc. 2021, 143, 17191. doi: 10.1021/jacs.1c07964.
- [7] A. Fawcett, A. Murtaza, C. H. U. Gregson, V. K. Aggarwal, J. Am. Chem. Soc. 2019, 141, 4573. doi: 10.1021/jacs.9b01513.
- [8] S. H. Bennett, A. Fawcett, E. H. Denton, T. Biberger, V. Fasano, N. Winter, V. K. Aggarwal, J. Am. Chem. Soc. 2020, 142, 16766. doi: 10.1021/jacs.0c07357.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
本文作者:孙苏赟
第四部分 Katsuki-Jacobsen不对称环氧化(Jacobsen AE)
Katsuki-Jacobsen不对称环氧化使用的手性salen类配体和锰的配合物对烯烃进行环氧化,常用的氧化剂有NaOCl,和高价碘试剂,例如PhI(OAc)2,等:

MnIII[(S,S/R,R)-salen]只是预催化剂,真正的催化剂是MnIV(O)配合物,NaOCl是化学计量使用的氧化剂:

这里值得注意的是,一般只限和顺式-1,2-二取代的双键反应,这也是其很大的一个局限。反应的立体面选择性是由配体的的影响造成的,其中叔丁基非常重要的大位阻基团,由于叔丁基的作用,烯烃不能从侧边靠近氧化剂,而只可能从前后方靠近,然而由于手性二胺的作用而控制了从底物只可以从配合物的后方靠近,其中二胺的构型决定了反应的实际面选择性,且使得反式烯烃很难参与该反应:

例如[1]:

端位双键的立体选择性大大降低:

但如过在反应中添加4-苯基吡啶氧化物,可以提高立体选择性,甚至可以使得反应在三取代的大位阻双键上发生很好的立体选择性环氧化:

关于反应的机理,目前尚有争议。主流的是有协同机理、自由基分步机理和环加成机理:

此外,Jacobsen的salen配体类催化剂从发明至今有多次的改进,并且在合成中展现了多种应用,这里是水解开环和叠氮化开环的两个应用:
(1) 配体的改进[2]:

引入不同的手性基团和增强其立体选择性,同时中心金属的不同电子密度可以使得此类催化剂可以适用于多种不同活性的底物。
(2) Co(III)催化的环氧化物水解开环动力学拆分(Hydrolytic kinetic resolution, HKR)

反应中加入水和醋酸是为了原位生成Co(III)物种:

一个例子[3]:

反应中的两个产物非常容易分离,可以轻易达到动力学拆分的目的。
反应的过程中,Co(III)可能会和产物环氧化物发生配合,然而只有R-环氧化物可以顺利在(S,S)-salen配体下发生这个过程,其对映异构体S-环氧化物无法参与其中去,因此造成了反应速率上的差异:

(3) Cr(III)催化的环氧化物叠氮化开环动力学拆分

例如[4]:

反应后处理,可以简单通过蒸发沸点较低的环氧化物而得到。
反应中的立体化学部分和HKR类似,但是反应的动力学研究发现反应速率是一个依Cr(III)浓度的二级反应。反应中最关键的是Cr(III)活化作用下的开环过程,其过程经历“头碰脚”的过渡态:jacs 1998 10780

References
- [1] Angew. Chem. Int. Ed. 1997, 36, 2060. Doi: 10.1002/anie.199720601
- [2] (a) Science 1997, 277, 936. DOI: 10.1126/science.277.5328.936. (b) J. Am. Chem. Soc. 2002, 124, 1307-1315. Doi: 10.1021/ja016737l. (c) J. Org. Chem. 1998, 63, 4876-4877. Doi: 10.1021/jo9810765. (d) Chem. Rev. 2005, 105, 1563-1602. Doi: 10.1021/cr0306945
- [3] Angew. Chem. Int. Ed . 2002, 41, 1374. Doi: 10.1002/1521-3773(20020415)41:8<1374::AID-ANIE1374>3.0.CO;2-8
- [4] J. Am. Chem. Soc. 1996, 118, 7420-7421. Doi: 10.1021/ja961708+
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文投稿作者 孙苏赟
研究背景
- 早期手性酮催化的不对称环氧化反应
1.1 手性酮催化的环氧化的起源
在上世纪八十年代,Curci发现了一种手性酮,它可以参与很多种烯烃的环氧化反应1,例如图中的这两种手性酮,但是他们使用时需要使用化学计量的量,最多可达到1/1,这些反应大多数需要在2-6°C进行,反应介质是DCM/H2O缓冲溶液的两相,同时作为相转移催化剂,产率在60%-90%之间,但是立体选择性很不好,大约只有10% e.e.。

1.2 在手性酮中引入吸电基团增加性能
探究发现,在手性酮的结构中若引入吸电集团,那么酮可以表现出更加优异的性能2,例如这样一个引入三氟甲基的酮参与反应的例子:

相同的,这些反应也是在两相系统中进行,但是他们的产率普遍要高一些,大概在80%,立体选择性也有微小的提升,大约为20%。
1.3 手性芳香酮催化反应
Brian S.Marples课题组在1995年报道了一例含氟的手性酮参与的环氧化反应3。这些芳香酮具有平面型的结构,并认为这样的平面型结构是有非常利于催化位阻较大的结构,例如发杂结构中的反式双键的,事实证明的确如此,而且这种芳香手性酮的催化效果很好。

虽然这些芳香手性酮参与的反应不能得到立体选择性非常高的产物,但是利用吸电基团对羰基进行活化的思路在之后的研究中被进一步的研究和开发了。
- 史一安的策略
在1996年,年轻的华裔化学家史一安报道了一种极其有用的反应,这个反应可以将烯烃原位氧化成环氧化合物,并且反应具有很好的立体选择性4。在这之后,这个反应也被叫做“史一安氧化反应”,或是叫“史一安环氧化反应”。即使是在今天,这个反应也经常在有机合成中被使用,用于高效的将烯烃转化成环氧化合物,而环氧化合物是一种活泼的反应中间体,可以接下来进行多种反应的转化。
2.1 反应方程

2.2 史一安对手性酮的设计

手性酮催化剂中包含的结构是为了实现以下的功能:
- 高效的反应底物发生联系,产生特定的化学选择性,因此反应中心附近结构的位阻需要较大以产生很好的化学选择性;
- 当多环化合物或是含有叔碳的化合物座位反应底物时,产生位阻的碳原子的构形可以根据反应设计的要求进行调控;
- 在立体化学上可以限制副反应的发生而产生副产物;
糖基手性酮的优点5:
- 碳水化合物作为基体的手性酮具有很好的立体构型和光学纯度,并且容易通过化学合成得到,价格低廉;
- 碳水化合物作为基体的手性酮是多取代的化合物,并且分子中含有氧原子,可以提高反应的化学活性;;
- 由于差向异构体的转化的影响,碳水化合物作为基体具有很好的分子构型,对于反应的立体选择性非常有利。
2.3 预催化剂的合成

这种手性酮及其对应异构体可以通过合成很简单的得到,而且反应的原料是D-fructose 和L-fructose,价格十分便宜,大约约15美元1千克。它们先通过和丙酮反应生成五元环,在再被PCC氧化得到酮。此外,在2003年,又有人报道了另一种用于从D-glucose合成含有氮元素的手性酮的新方法。
2.4 手性酮催化剂的结构7-9

手性酮上的取代基和取代基的性质与酮的催化效果有很大的关系,酮A具有五元环螺六元环的结构,它的性能较六元环螺六元环的酮B的催化效果更好,特别是参与反式二取代的双键的反应时,可以表现出更好的效果;而对于C和D,他们结构中的非环状的结构对于立体化学的控制能力不如前两者的出色。
酮A具有很好的立体选择性,因为五元螺环的缩酮结构可以有力的控制反应的过渡态,使得反应通过螺状都过渡态进行,相同类似的基团在B,C和D中也存在,但是对于螺形过渡态的控制不如A,因此立体选择性也较弱一些。
对于E来说,它可以从(-)-qunic acid合成,但是E的结构和前者有很大的不同之处,尽管如此,E的催化效果一般,34%-95%产率,52%-94% e.e。
2.5 环氧化的反应机理
环氧化过程主要的两个极限过渡态是平面型过渡态和螺型过渡态10。

过氧丙酮进行环氧化反应时发现,顺式双键的反应活性一般要高于反式双键,可能的原因是此类反应是通过螺型过渡态进行的。相同地,史一安环氧化反应的过程中,手性酮会经历一个过氧丙酮类似物的中间体,这个中间体再对烯烃进行环氧化反应,反应过程中可能是螺型过渡态,但是通常时候是平面型过渡态占主导,因为普遍认为螺型过渡态是电子丰时的过渡态,因为氧原子孤对电子和烯烃的π*轨道只见的相互作用,但是史一安的环氧化反应中底物越缺电子则越有利于反应的发生,因此更加可能是平面型过渡态发生反应;此外,反应的产物大多是(R,R)-构型的,这更加支持了反应的过渡态的理论。

在反应中,真正的氧化剂是具有氧化性的被活化的手性过氧化酮,这个过氧酮的中间体是通过手性在原位被Oxone氧化而得到的,而这个中间体可以看做是过氧丙酮的类似物,之后这样的过氧丙酮类似物和烯烃发生环氧化反应得到环氧化物的产物。但是反应会存在一个竞争的反应,即反应中会发生Baeyer-Villiger重排,也就是Baeyer-Villiger氧化反应,这个副反应会严重的影响底物的转化率。此外,在这个反应中,只有反式双键可以得到具有较高立体选择性的产物,这样可以和只能和顺式烯烃反应的Jacobsen氧化反应和Sharpless AE作为相互补充的反应。但是史一安反应中的一个缺点是催化的手性酮的用量较大,其反应中的浓度需要达到20-30 mol%。
参考文献
- J. Chem. Soc., Chem. Commun., 1984, 155-156. , DOI:10.1039/C39840000155
- Tetrahedron, 1995, 36(32), 5831-5834, DOI:1016/0040-4039(95)01108-T
- Tetrahedron, 1995, 51(12), 3587-3606, DOI:10.1016/0040-4020(95)00075-J
- Tetrahedron, 1991, 47(12), 2133-2144, DOI:1016/S0040-4020(01)96124-1
- J. Am. Chem. Soc., 1997, 119, 11224-11235, DOI:10.1021/ja972272g
- J. Org. Chem., 1997, 62, 8622-8623, DOI:10.1021/jo971701q
- J. Org. Chem., 1998,63, 8475-8485, DOI:10.1021/jo9817218
- J. Org. Chem., 1999, 64, 6443-6458, DOI:10.1021/jo9908849
- J. Am. Chem. Soc., 1996, 118, 9806-9807, DOI:10.1021/ja962345g
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
金沢大学的国嶋课题组开发出了新型环氧化试剂2-hydroperoxy-4,6-diphenyl-1,3,5-triazine (Triazox)、对于其有用性的报道近期发表在了Org. Lett.上。
“An Isolable and Bench-Stable Epoxidizing Reagent Based on Triazine: Triazox”
K. Yamada, Y. Igarashi, T. Betsuyaku, M. Kitamura, K. Hirata, K. Hioki, M. Kunishima*, Org. Lett., ASAP.
DOI: 10.1021/acs.orglett.8b00560
烯烃的环氧化反应
作为烯烃的常见环氧化试剂,大致有如下几种。
- H2O2
- mCPBA
- DMDO
- Triphenylsilyl hydroperoxide
对于环氧化的反应机理,有如下3种。
①对于含有吸电子基团(共轭)的底物,过氧酸阴离子的共轭加成后,再发生对氧的分子内亲核进攻形成环氧基团。(具体参考本网站反应库:亲核环氧化反应)
②协同式反应(具体参考本网站反应库:{mCPBA环氧化、过氧化酮氧化、夏普莱斯-香月不对称环氧化反应、史不对称环氧化})
③自由基型反应(具体参考本网站反应库:Jacobsen-Katsuki Epoxidation{对于该反应的机理还存在一些争议})
另一方面,每种方法都有在制备容易,试剂价格低,试验稳定性和安全性这些方面有其长处短处。
这一次金沢大学的国嶋课题组,开发出了标题图中的2-hydroperoxy-4,6-diphenyl-1,3,5-triazine (Triazox)环氧化试剂,该试剂制备简单,纯化度高,安全性,反应温和等长处。
试剂制备法

作者其实最初以2-氯-4,6-二甲氧基-1,3,5-三嗪(CDMT)为原料,合成三嗪化环氧化试剂,然而因为该试剂吸湿性高,容易坏。
为了改善这个问题,最后作者使用能CDPT合成了Triazox(如上图所示)。该试剂如m-CPBA一样,可以制备成无水晶体,且本身安全性很高。
与其他环氧化试剂的对比
作者在表格中清楚地显示了该试剂的有用性/特性。下面是对其中的一部分描述的整理(详情请参阅论文)。
其他环氧化试剂的缺点用蓝色标出,Triazox的优势特性用红色表示(m-CPBA由于可市售,没有用颜色标出)。

从表中可以看出,Triazox的特性是高纯度试剂,可以说作为环氧化试剂的可靠性很高。
另外,coproduct的酸性比较低也是Triazox的一大优点,相对的常用的m-CPBA的coproduct酸性很强,这样对于对酸敏感的底物就比较吃力了。
底物的适用性
作者在筛选了溶剂后,又进行了底物适用性的研究
底物的适用性(引用自原论文)

从上图底物适用性范围可以看出覆盖的底物范围还是很广的,因此可以说Triazox的有用性还是相当不错的。关于环己烯醇的非对映选择性,由于邻位基的参与显示出一定的选择性,详细请参考Supporting Information。
对于本反应,一打有点是,对于反应的纯化来说,省去了常规的分液萃取操作。反应完后仅仅需要过滤,浓缩,过柱即可。
结尾
最后,对于该试剂今后广泛的使用性的期待,引用下SI中该论文作者的一段话来结束本记事。有兴趣的可以读一下。
Caution.
Triazox is not detonated by a hammer blow. Measurement of self-accelerating decomposition temperature and flash point, and thermal stability test were not conducted. In our laboratory, Triazox is stored at refrigerator and epoxidation was conducted in the test tube reinforced by polyolefin shrink wrap film.
Although we experienced no hazards in the course of this work, routine precautions (shields, fume hoods) should be conducted whenever possible, and the reaction should be run first on a small scale, as organic peroxides are intrinsically explosive.引用SI
参考文献
- N. N. Schwartz, J. H. Blumbergs, J. Org. Chem., 1964, 29, 1976.
DOI: 10.1021/jo01030a078 - R. W. Murray, Chem. Rev. 1989, 89, 1187.
DOI: 10.1021/cr00095a013
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
史氏环氧化反应 (Shi Epoxidation)是华裔化学家史一安所发明的一种不对称环氧化反应(目前已经全职回到南京大学工作)。该反应的氧化剂是过一硫酸钾(KHSO5),催化剂是果糖的衍生物。史氏环氧化反应由于在冰浴-室温这种温和条件下进行,而且适用范围广泛,反式二取代烯与三取代烯均可作为反应的底物,因此成为有机合成的重要工具之一。略不足的是,催化剂用量稍多(20-30mol%)。
新利官方网站
基本文献
- Tu, Y.; Wang, Z.-X.; Shi, Y. J. Am. Chem. Soc. 1996, 118, 9806. DOI: 10.1021/ja962345g
- Wang, Z.-X.; Tu, W; Frohn, M.; Zhang, J.-R.; Shi, Y. J. Am. Chem. Soc. 1997, 119, 11224.doi:10.1021/ja972272g
- Frohn, M.; Shi, Y. Synthesis 2000, 14, 1979. DOI: 10.1055/s-2000-8715
- Adam, W.; Saha-Moeller, C. R.; Zhao, C. -G. Org. React. 2002, 61, 219.
- Shi, Y. Acc. Chem. Res. 2004, 37, 488. DOI: 10.1021/ar030063x
反应机理
体系中生成的手性dioxirane是实际上的活性种。该反应需要在碱性条件下进行,如果是酸性条件的话,会产生副反应Baeyer-Villiger氧化使催化剂失活。

反应实例
对于含有多个双键的底物,利用本方法也可以保证立体控制的同时,一次性进行环氧化。
Glabrescol的合成[1]

级联反应合成聚醚化合物[2]

Octalacin的合成研究[3]:在含有共轭二烯的情况下,可以发生富电子或空间位阻小的烯烃进行选择性单环氧化。

实验步骤
实验技巧
参考文献
[1] Xiong, Z.; Corey, E. J. J. Am. Chem. Soc. 2000, 122, 4831, 9328. DOI: 10.1021/ja000842y DOI: 10.1021/ja0024901[2] Simpson, G. L.; Heffron, T. P.; Merino, E.; Jamison, T. F. J. Am. Chem. Soc. 2006, 128, 1056. DOI: 10.1021/ja057973p [3] Bluet, G.; Campagne, J.-M. Synlett 2000, 221. DOI: 10.1055/s-2000-6491
相关反应
- Swern Oxidation
- Rubottom Oxidation
- Jacobsen-Katsuki Epoxidation
- Nucleophilic Epoxidation with Peroxide
- Davis Oxidation
- Prilezhaev Epoxidation
- Sharpless-Katsuki Asymmetric Epoxidation (Sharpless AE)
外部链接
- Shi Epoxidation (Wikipedia)
- Yian Shi Group
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
- 概要
丙酮与Oxone反应配制的甲基二环氧乙烷(DMDO),可以用于烯烃的环氧化反应。另一种过氧化酮甲基三氟甲基二环氧乙烷(TFDO)的反应性是DMDO的600倍。

与mCBPA相比,该反应具有以下优点:1.反应可在中性条件下进行,2.反应中只生成丙酮副产物,后处理比较简单,3.氧化剂自身廉价易得。但是,该氧化试剂由于稳定性比较差,所以得现配现用。另外还必须在减压蒸馏的条件下配制,操作方面也比较麻烦。
对于特定的底物,可以对其中的双键进行环氧化得到环氧化物产物。
- 基本文献
- Murray, R. W.; Jayaraman, R. J. Org. Chem. 1985, 50, 2847. DOI: 10.1021/jo00216a007
- Adam, W.; Chan, Y.-Y.; Cremer, D.; Gauss, J.; Scheutzow, D.; Schindler, M. J. Org. Chem. 1987, 52, 2800. DOI: 10.1021/jo00389a029
- Mello, R.; Fiorentino, M.; Sciacovelli, O.; Curci, R. J. Org. Chem. 1988, 53, 3890. DOI:10.1021/jo00251a053
- Mello, R.; Fiorentino, M.; Fusco, C.; Curci, R. J. Am. Chem. Soc. 1989, 111, 6749. DOI:10.1021/ja00199a039
- Adam, W.; Hadjiarapoglou, L.; Nestler, B. Tetrahedron Lett. 1990, 31, 331. doi:10.1016/S0040-4039(00)94547-7
- Yang, D.; Wong, M.-K.; Yip. Y.-C. J. Org. Chem. 1995, 60, 3887. DOI: 10.1021/jo00117a046
- Frohn, M.; Wang, Z.-X.; Shi, Y. J. Org. Chem. 1998, 63, 6425. DOI: 10.1021/jo980604+
- Yang, D.; Wong, M.-K.; Wang, X. -C.; Tang, Y.-C. J. Am. Chem. Soc. 1998, 120, 6611. DOI:10.1021/ja980916u
- Ferraz, H. M. C.; Muzzi, R. M.; Vieira, T. O.; Viertler, H. Tetrahedron Lett. 2000, 41, 5021. doi:10.1016/S0040-4039(00)00769-3
<review>
- Murray, R. W. Chem Rev. 1989, 89, 1187. DOI: 10.1021/cr00095a013
- Curci, R.; Dinoi, A.; Rubino, M. F. Pure Appl. Chem. 1995, 67, 811. doi: 10.1351/pac199567050811
- Curci, R.; D’Accolti, L.; Fusco, C. Acc. Chem. Res. 2006, 39, 1. DOI: 10.1021/ar050163y
- Adam, W.; Saha-Moller, C. R.; Zhao, C.-G. Org. React. 2002, 61, 219.
- 反应机理

- 反应实例
Morphine的合成[1]

Merrilactone A的合成[2]

选择性C-H氧化实例[3]: 甲基的反应性比亚甲基高。

Bryostatin Analogue的合成应用[4]

- 实验步骤
甲基二环氧乙烷的配制[5a]

在装有搅拌子的三颈瓶中加入蒸馏水(120mL)、丙酮(100mL)、碳酸氢钠(45g),在冰浴下剧烈搅拌5分钟。分三次在该溶液中加入Oxone(90g),保持冰浴状态,搅拌15~20分钟。生成的DMDO通过减压蒸馏纯化获得(使用50-120 mmHg的简易泵、1-2h)。接收瓶的话需要保持在-78℃,并且在瓶中需要加入4A的分子筛以达到除水的目的。DMDO的浓度可以用茴香硫醚NMR滴定法测定。
- 实验技巧
※生成过氧化物的反应通常有爆炸的危险性,所以需要配备防爆挡板。
※DMDO具有挥发性,所以反应需要在通风橱中进行。
※由于该氧化剂的氧化反应性很强,所以绝对要避免接触到皮肤或者吸入呼吸道。所以必须得佩戴防护手套,但是不能是橡胶质地的。
- 参考文献
[2] (a) Inoue, M.; Sato, T.; Hirama, M. J. Am. Chem. Soc. 2003, 125, 10772. DOI: 10.1021/ja036587+ (b) Inoue, M.; Sato, T.; Hirama, M. Angew. Chem. Int. Ed. 2006, 45, 4843. doi:10.1002/anie.200601358 (c) Inoue, M.; Lee, N.; Kasuya, S.; Sato, T.; Hirama, M.; Moriyama, M.; Fukuyama, Y. J. Org. Chem. 2007, 72, 3065. DOI:10.1021/jo0700474
[3] (a) Bovicelli, P.; Lupattelli, P.; Mincione, E.; Prencipe, T.; Curci, R. J. Org. Chem. 1991, 57, 2182. DOI:10.1021/jo00033a053 (b) Iida, T.; Yamaguchi, T.; Nakamori, R.; Hikosaka, M.; Mano, N.; Goto, J.; Nambara, T. J. Chem. Soc. Perkin Trans. 1 2001, 2229. DOI: 10.1039/B104938K
[4] Wender, P. A.; Hilinski, M. K.; Mayweb, A, V, W, Org. Lett. 2005, 7, 79. DOI: 10.1021/ol047859w
[5] (a) Li, Y.; Tang, P.; Chen, Y.; Yu, B. J. Org. Chem. 2008, 73, 4323. DOI: 10.1021/jo8003875 (b) Murray, R. W.; Singh, M. Org. Synth. Coll. Vol. 9, 288 (1988). [website]
- 概要
该反应是通过硫鎓盐进行的1,2-或1,4-加成反应。对于羰基的1,2-加成反应得到的是过氧化物,而对电子不足的烯烃进行共轭加成则是得到环丙烷产物。
- 基本文献
・ Corey, E. J.; Chaykovsky, M. J. Am. Chem. Soc. 1962, 84, 867. doi:10.1021/ja00864a040
・ Corey, E. J.; Chaykovsky, M. J. Am. Chem. Soc. 1965, 87, 1353. doi:10.1021/ja01084a034
・ Corey, E. J.; Chaykovsky, M. Org. Synth. 1969, 49, 78. [website]
・ Review: Aggarwal, V. K.; Richardson, J. Chem. Commun. 2003, 2644. DOI:10.1039/b304625g
- 反应机理
硫原子相对于磷(Wittig反应)或硼(Peterson反应)来说对氧的亲和性比较弱,所以羰基更容易与硫鎓盐反应得到环氧化物。另一方面,亚砜形式的硫鎓盐相对来说更加稳定,所以即使进行1,2-加成后,其反应方向更容易向着其逆反应进行,最终主要形成了1,4-附加产物(环丙烷化合物)。

- 反应实例
近年MacMillan等人用、不对称有机催化剂、开发出了不对称Corey-Chaykovsky环丙烷化反应。[1]

Phyllanthocin的合成[2]

Batrachotoxinin A的合成[3]

- 实验步骤
- 实验技巧
- 参考文献
[2] Smith, A. B., III; Fukui, M.; Vaccaro, H. A.; Empfield, J. R. J. Am. Chem. Soc. 1991, 113, 2071. DOI: 10.1021/ja00006a029
[3] Kurosu, M.; Marcin, L. R.; Grinsteiner, T. J.; Kishi, Y. J. Am. Chem. Soc. 1998, 120, 6627.doi:10.1021/ja981258g
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
- 概要
硅烯醇醚在mCPBA或二环氧乙烷中被环氧化后,能够快速进行重排,得到α-硅氧基酮。该反应经常被用于在α位选择性导入羟基。
- 基本文献
・ Rubottom, G. M.; Vazquez, M. A.; Pelegrina, D. R. Tetrahedron Lett. 1974, 15, 4319. doi:10.1016/S0040-4039(01)92153-7
・ Brook, A. G.; Macrae, D. M. J. Organomet. Chem. 1974, 77, C19. doi:10.1016/S0022-328X(00)81332-7
・ Hassner, A.; Reuss, R. H.; Pinnick, H. W. J. Org. Chem. 1975, 40, 3427. DOI: 10.1021/jo00911a027
- 反应机理

- 反应实例
(-)-Stenine的合成[1]

- 实验步骤
- 实验技巧
- 参考文献
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
- 概要
缺电子的烯烃,在碱性条件下,被双氧水与过氧化叔丁醇环氧化的反应。由于该反应是亲核反应,所以底物中没有吸电子基团的话,环氧化无法进行。
- 基本文献
・Julia, S.; Masana, J.; Vega, J. Angew. Chem. Int. Ed., Engl. 1980, 19, 929. doi:10.1002/anie.198009291
・Banfi, S.; Colonna, S.; Molinari, H.; Julia Guixer, J. Tetrahedron 1984, 40, 5207. doi:10.1016/S0040-4020(01)91272-4
・House, H. O.; Ro, R. S. J. Am. Chem. Soc. 1958, 80, 2428. DOI: 10.1021/ja01543a021
- 反应机理
亲核1,4-加成→羟基的脱除。该反应机理与mCPBA氧化不同,必须注意。

- 反应实例
有机催化剂参与的α,β-不饱和醛的不对称环氧化反应[1]

化学选择性的例子[2]:该条件下异戊烯不会被环氧化。

- 实验步骤
- 实验技巧
- 参考文献
[2] Grieco, P. A.; Nishizawa, M.; Oguri, T.; Burke, S. D.; Marinovic, N. J. Am. Chem. Soc. 1977, 99, 5773. DOI: 10.1021/ja00459a039
- 概要
利用过氧化物把烯烃转化为环氧化物的反应。其中使用的间氯过氧苯甲酸(mCPBA)是固体,而且相对来说不易爆炸、所以经常在实验室中被使用。二甲基过氧化酮等也可以被用于此反应。
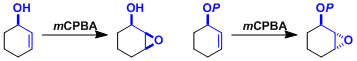
一般来说,富电子的烯烃反应性更高: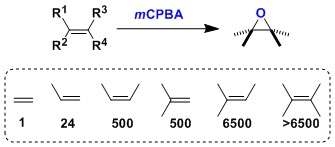 如果邻位存在无保护的羟基、由于反应中氢键的影响,会形成非对映选择性的过氧化产物(Henbest法则)。相对的,羟基被保护的情况下那么就会形成相反的立体选择性的环氧化产物。
如果邻位存在无保护的羟基、由于反应中氢键的影响,会形成非对映选择性的过氧化产物(Henbest法则)。相对的,羟基被保护的情况下那么就会形成相反的立体选择性的环氧化产物。
- 基本文献
・Prilezhaev, N. Ber. 1909, 42, 4811. doi:10.1002/cber.190904204100
・Swern, D. Org. React. 1953, 7, 378.
・Henbest Rule: Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307. DOI: 10.1021/cr00020a002
- 反应机理
反应是通过熟知的“蝴蝶机理”进行。烯烃保持原来的立体构型不变,烯烃上的给电子基团能够促进该反应的进行。 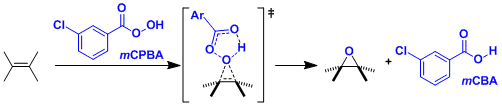
- 反应实例
- 实验步骤
- 实验技巧
- 参考文献