概要
使用腈的苯胺衍生物的O-酰化反应。 该反应中,苯胺无需事先保护就行进行。
众所周知,对于苯胺的Friedel-Crafts酰基化,由于氮原子上的孤对电子对会与路易斯酸发生配位,导致芳香环的电子密度下降,使得反应很难进行。
而本反应通过使用两种路易斯酸,回避了上述问题。
通常的路易斯酸的组合是BCl3-AlCl3、而使用与氯原子亲和性更高的GaCl3,可以使得反应在更加温和的条件下进行。
基本文献
・Sugasawa, T.; Toyoda, T.; Adachi, M.; Sasakura, K. J. Am. Chem. Soc. 1978, 100, 4842. DOI: 10.1021/ja00483a034
・Douglas, A. W.; Abramson, N. L.; Houpis, I. N.; Karady, S.; Molina, A.; Xavier, L. C.; Yasuda, N. Tetrahedron Lett. 1994, 35, 6807. doi:10.1016/0040-4039(94)85010-0
・Prasad, K.; Lee, G. T.; Chaudhary, A.; Girgis, M. J.; Streemke, J. W.; Repic, O. Org. Proc. Res. Dev. 2003, 7, 723. DOI: 10.1021/op0340659
反应机理
参考:Tetrahedron Lett.1994, 35, 6807.

反应实例
使用菅沢反应进行的吲哚合成[1]

参考文献
[1] Pei, T.; Tellers, D. M.; Streckfuss, E. C.; Chen, C.-y.; Davies, I. W. Tetrahedron2009, 65, 3285. doi:10.1016/j.tet.2008.11.026
外部链接
・Lewis Acid Catlaysis – Wikipedia
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
此反应由Meinwald在1963年首次报道,该反应为路易斯酸促进下的环氧化物转化为羰基化合物(酮或醛)的反应。除了路易斯酸,还发现质子酸也适合于这种重排。该反应通常是在无水条件以及当量或者过量的路易斯酸存在下,在惰性气氛保护下进行的。产物由环氧部分连接的取代基迁移性质、所用的溶剂和路易斯酸共同决定。
基本文献
- Meinwald, J.; Labana, S. S. and Chadha, M. S., J. Am. Chem. Soc.1963, 85, 582. DOI: 10.1021/ja00888a022
反应机理[1]

反应实例
- (S)-cuparene的合成[2]:
 2. 3-arylquinolin-4(1H)-ones的构建[3]:
2. 3-arylquinolin-4(1H)-ones的构建[3]:

实验步骤[4]

向35mg环氧化物(0.11mmol)的3mL苯溶液中加入117mg无水溴化锌(0.52mmol),将混合物于70℃下搅拌反应10分钟。冷却至室温后,将混合物用乙醚稀释,用水和盐水洗涤,用无水MgSO4干燥。除去溶剂后,残余物通过柱色谱法纯化。最终得到27mg产物,为无色油状物,产率为80%。
参考文献
- Kita, Y.; Kitagaki, S.; Yoshida, Y.; Mihara, S.; Fang, D.-F.; Kondo, M.; Okamoto, S.; Imai, R.;Akai, S. and Fujioka, H., J. Org. Chem. 1997, 62, 4991. DOI: 10.1021/jo970035q
- Kumar, R.; Halder, J.; Nanda, S. Tetrahedron, 2017, 73(6), 809-818 .DOI:1016/j.tet.2016.12.072
- Wang, S.; Zhao, C.; Liu, T. et al. Tetrahedron, 2016, 72(44): 7025-7031. DOI:1016/j.tet.2016.09.039
- Li W.-D. Z;. and Yang, Y.-R. Org. Lett. 2005, 7, 3107. DOI:10.1021/ol051141e
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
以可以纯化保存的氟化糖作为糖基化的donor进行的糖基化的手法。最初向山等人利用含锡的路易斯酸作为活化剂活化进行,之后铃木等人改进了其方法,通过使用含正电性的锆或铪络合物等hard性的路易斯酸,提高了反应活性,并且扩大了反应的适用范围。
LiClO4或者强质子酸(TfOH等)也可能活化氟化糖。
基本文献
- Mukaiyama, T.; Murai, Y.; Shoda, S. Chem. Lett. 1981, 431. doi:10.1246/cl.1981.431
- Matsumoto, T.; Maeta, H.; Suzuki, K.; Tsuchihashi, G. Tetrahedron Lett.1988, 29, 3567. doi:10.1016/0040-4039(88)85294-8
- Suzuki, K.; Maeta, H.; Matsumoto, T.; Tsuchihashi, G. Tetrahedron Lett.1988, 29, 3571. doi:10.1016/0040-4039(88)85295-X
- Suzuki, K.; Maeta, H.; Matsumoto, T. Tetrahedron Lett. 1989, 30, 4853. doi:10.1016/S0040-4039(01)80526-8
<review>
- 鈴木啓介、長澤徹哉 有機合成化学協会誌 1992, 50, 378. doi:10.5059/yukigoseikyokaishi.50.378
- Toshima, K.; Tatsuta, K. Chem. Rev. 1993, 93, 1503. DOI: 10.1021/cr00020a006
- Mukaiyama, T. Tetrahedron 1999, 55, 8609. doi:10.1016/S0040-4020(99)00437-8
- Mukaiyama, T. Angew. Chem. Int. Ed. 2004, 43, 5590. DOI: 10.1002/anie.200300641
- Zhu, X.; Schmidt, R. R. Angew. Chem. Int. Ed. 2009, 48, 1900. DOI: 10.1002/anie.200802036
反应机理
与其他卤代糖相比氟化的糖的热/化学稳定性最高。这是由于C-F结合键的能量的大小决定的。(C-F: 552 kJ/mol, C-Cl: 397 kJ/mol, C-Br: 280 kJ/mol)。也是由于该稳定性,所以氟化糖是可以通过过柱子纯化的。
反应实例
Benanomicin B的全合成[1]

Gilvocarcin M的全合成[2]:苯酚作为糖基化的acceptor,通过在高温下O→C重排得到C-糖基化产物。

实验注意点
反应中如果有使用到高氯酸银,虽然活性很好,但是具有潜在爆炸性。最好先用三氟甲磺酸银试一下反应,如果能够进行的话就用比较安全的三氟甲磺酸银进行活化。
参考文献
- (a) Ohmori, K.; Tamiya, M.; Kitamura, M.; Kato, H.; Oorui, M.; Suzuki, K. Angew. Chem. Int. Ed. 2005, 44, 3871. DOI: 10.1002/anie.200501210 (b) Tamiya, M.; Ohmori, K.; Kitamura, M.; Kato, H.; Arai, T.; Oorui, M.; Suzuki, K. Chem. Eur. J. 2007, 13, 9791. DOI: 10.1002/chem.200700863
- (a) Matsumoto, T.; Hosoya, T.; Suzuki, K. J. Am. Chem. Soc. 1992, 114, 3568. DOI: 10.1021/ja00035a069 (b) Hosoya, T.; Takashiro, E.; Matsumoto, T.; Suzuki, K. J. Am. Chem. Soc. 1994, 116, 1004. DOI: 10.1021/ja00082a023
关联图书

Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance
- 参考価格¥ 25,866
価格¥ 25,584(2016/12/14 21:54時点) - 出版日2008/03/31
- 商品ランキング269,473位
- ハードカバー524ページ
- ISBN-103527317805
- ISBN-139783527317806
- 出版社Wiley-VCH

Reactivity Tuning in Oligosaccharide Assembly (Topics in Current Chemistry)
- 参考価格¥ 47,913
価格¥ 47,545(2016/12/15 11:12時点) - 出版日2013/07/15
- ペーパーバック296ページ
- ISBN-103642268242
- ISBN-139783642268243
- 出版社Springer
外部相关链接
- Chemical Glycosylation – Wikipedia
- Glycosylation – Wikipedia
- Armed and disarmed saccharides – Wikipedia
- グリコシル化 – Wikipedia
- グリコシル化反応(TCI)
- Glycosylation(Sigma-Aldrich)
- グリコシル化反応(メタロセン法)
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
β-酮酸甲酯与LiCl在DMSO中加热,在基本中性条件下引发脱碳酸的反应。该方法常常用于乙酰乙酸酯/丙二酸酯合成中。
基本文献
- Krapcho, A. P.; Glynn, G. A.; Grenon, B. J. Tetrahedron Lett. 1967, 8, 215. doi:10.1016/S0040-4039(00)90519-7
- Krapcho, A. P.; Jahngen, E. G. E.; Lovey, A. J.; Short, F. W. Tetrahedron Lett. 1974, 15, 1091. doi:10.1016/S0040-4039(01)82414-X
- Krapcho, A. P.; Weimaster, J. F.; Eldridge, J. M.; Jahngen, E. G. E.; Lovey, A. J.; Stephens, W. P. J. Org. Chem. 1978, 43, 138. doi:10.1021/jo00395a032
- review: Krapcho, A. P. Synthesis 1982, 805. DOI: 10.1055/s-1982-29953
- review: Krapcho, A. P. Synthesis 1982, 893. DOI: 10.1055/s-1982-29991
反应机理
LiCl作为路易斯酸活化底物,然后甲酯一侧发生SN2反应完成脱碳酸的过程。(参照:Tetrahedron 1990, 46, 3929)

反应实例
实验步骤
实验技巧
参考文献
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
概要
氯化铝等路易斯酸催化下,氢氰酸与芳香族化合物(如苯)作用生成芳香醛的反应。
这个反应。。。狠毒。。。=。=
基本文献
- Gattermann, L. Ber. 1898, 31, 1149.
- Gattermann, L. Ann. 1907, 313.
- Truce, W.E. Org. React. 1957, 9, 37.
- Sato, Y.; Yato, M.; Ohwada, T.; Saito, S.; Shudo, K. J. Am. Chem. Soc. 1995, 117, 3037. DOI:10.1021/ja00116a009
反应机理
很明显的亲电取代机理

反应实例
实验步骤
实验技巧
参考文献
- L. Gattermann. 1890, 23 (1): 1218–1228.doi:10.1002/cber.189002301199.
- L. Gattermann, W. Berchelmann. 1898, 31 (2): 1765–1769.doi:10.1002/cber.18980310281.
- 概要
叠氮与羰基化合物在路易斯酸的存在家发生重排,最终形成相同碳数酰胺的反应。
该反应经常被用于通过分子内反应合成生物碱骨架。
- 基本文献
- Boyer, J. H.; Hamer, J. J. Am. Chem. Soc. 1955, 77 , 951. doi:10.1021/ja01609a045
- Aube, J.; Milligan, G. L. J. Am. Chem. Soc. 1991, 113, 8965. DOI: 10.1021/ja00023a065
- Aube, J.; Milligan, G. L.; Mossman, C. J. J. Org. Chem. 1992, 57, 1635. DOI: 10.1021/jo00032a003
- Gracias, V.; Milligan, G. L.; Aube, J. J. Am. Chem. Soc. 1995, 117, 8047. DOI: 10.1021/ja00135a036
- Gracias, V.; Frank, K. E.; Milligan, G. L.; Aube, J. Tetrahedron 1997, 53, 16241. doi: 10.1016/S0040-4020(97)01012-0
- Desai, P.; Schildknegt, K.; Agrios, K. A.; Mossman, C.; Milligan, G. L.; Aube, J. J. Am. Chem. Soc.2000, 122, 7226. DOI: 10.1021/ja000490v
- Grecian,S.; Aube, J. Org. Synth. 2007, 84, 347. [website]
- 反应机理

- 反应实例
Stenine的全合成[1]

- 实验步骤
- 实验技巧
- 参考文献
山本 尚(Yamamoto Hisashi 1943年7月16日- )是日本的有机化学家、工学博士。日本中部大学教授。以lewis酸的研究为中心开发了很多有用的有机合成催化反应。日本海外杰出的化学家之一。
经历
1943年7月16日出生 。京都大学工学部工业化学科毕业后(野崎一教授)赴美留学、师从哈佛大学E.J.Corey教授门下取得博士学位。之后在京都大学担任助手、又在夏威夷大学、之后成为名古屋大学教授。退休后任职芝加哥大学化学科的教授,现在是名古屋中部大学教授。
- 1967 京都大学工学部工业化学科 卒業 (野崎一 教授)
- 1971 哈佛大学 Ph.D (E. J. Corey教授)
- 1971 东丽 Toray Industries, Inc. (辻 二郎 教授)
- 1972 京都大学 助手
- 1976 京都大学 讲师
- 1977 夏威夷大学 准教授
- 1980 名古屋大学 助教授
- 1983 名古屋大学 教授
- 2002 芝加哥大学化学科 Arthur Holly Compton Distinguished Professor
- 2011 中部大学教授
- 2016 日本化学会会长
获奖经历
1977 日本化学会进步奖
1988 IBM科学奖
1993 Prelog Medal
1995 日本化学会学会奖
1997 东丽科学技术奖
1998 Max-Tishler Prize
2002 紫綬褒奖章
2003 Molecular Chirality Award
2004 Yamada Prize
2006 Tetrahedron Prize
2007 日本学士院奖
2007 洪堡奖
2008 有机合成化学协会 特別奖
2009 ACS Award for Creative Work in Synthesitc Organic Chemistry
2011 野依奖
2012 藤原奖
研究
・手性路易斯酸的不对称催化反应的研究
・研究用路易斯酸做催化剂的酰化、酯化反应
・路易斯酸复合型手性Brønsted酸的开发

・用MAD、Aluminum Tris(2,6- diphenylphenoxide(ATPH)的反应

・使用超大位阻的试剂的有机合成反应的开发
关联论文
- [1] (a) Yamamoto, H.; Maruoka, K. Pure & Appl. Chem. 1988, 760, 21. (b) Yamamoto, H.; Saito, S. Pure & Appl. Chem. 1999,71, 239. [PDF]
- [2] Yamamoto, H.; Futatsugi, K.; Angew. Chem. Int. Ed., 2005, 44, 1924. DOI: 10.1002/anie.200460394
- [3] (a) Furuta, K.; Miwa, Y.; Iwanaga, K.; Yamamoto, H. J. Am. Chem. Soc. 1988, 110, 6254. DOI:10.1021/ja00226a055 (b) Ishihara, K.; Gao, Q.; Yamamoto, H. J. Am. Chem. Soc. 1993, 115, 10412. DOI: 10.1021/ja00075a088
- [4] Yamamoto, H., M. Kawasaki, Bull. Chem. Soc. Jpn., 2007, 80, 595. DOI: 10.1246/bcsj.80.595
- [5] Abell, J.P.; Yamamoto, H. Chem. Soc. Rev. 2010, 39, 61. DOI:10.1039/B907303P
评论&其他
- 山本教授在名古屋大学满年岁退休后、去芝加哥大学任教。是向海外流动的高级人才中的一员。日本国内对大学教员有退休年龄限制(通常60岁)、满年龄后就没办法招收研究员和学生,对于那些依然有精力、想继续从事研究的教授是一种限制。相反,在美国,即使年龄超过80岁,也还有继续从事科研的教授,这也是山本教授选择去芝加哥大学的理由。
- 即使是近几年,使用手性路易斯酸催化剂的羟醛缩合反应,α-羟胺化反应、亚硝基羟醛-迈克尔反应、NHK反应、樱井-细见反应等不对称反应为中心的论文,每年都多达十篇以上。
- 研究室的主页中发表工作一项中,每项课题旁都附有该学生的照片和简介。这样做也能引起人们去更好的关注。
- 山本教授曾与万有财团协作,致力于许多针对有机合成化学领域的年轻研究者的育成计划。最开始以Merk-Banyu Lectureship award (MBLA), 是熟为人知的40岁以下研究者的最高荣誉奖赏、以及早期的Banyu Chemist Award之后,到现在针对研究生都可以申请的大津会议等一系列鼓励年轻研究者的活动。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
- 概要
烯丙基硅参与的烯丙基化反应。烯丙基硅试剂是相对烯丙基锡毒性低、比烯丙基格利雅试剂及锂试剂稳定的有机合成试剂。
常加入Lewis酸,氟化物来推动反应进行。特别是,在lewis酸作用下,烯丙基格氏试剂,烯丙基锂试剂按γ位選択进行、根据底物不同能达到很好的立体选择性。还有共轭烯醇的烯丙基化反应只按照共轭加成的方式进行。
- 基本文献
・Hosomi, A.; Endo, M.; Sakurai, H. Chem. Lett. 1976, 941. doi:10.1246/cl.1976.941
・Hosomi, A.; Sakurai, H. Tetrahedron Lett. 1976, 17, 1295. doi:10.1016/S0040-4039(00)78044-0
・Hosomi, A.; Sakurai, H. J. Am. Chem. Soc. 1977, 99, 1673. DOI: 10.1021/ja00447a080
・Wilson, S. R.; Price, M.F. J. Am. Chem. Soc. 1982, 104, 1124. DOI: 10.1021/ja00368a049
・Review: Fleming, I. et al. Org. React. 1989, 37, 57.
・Review: Fleming, I. Comprehensive Organic Syntheis 1991, 2, 563.
- 反应机理
烯丙基β位的碳正离子是因超共轭的原因,较稳定。在此基础上提出以下的反应机理。硅的lewis酸性较弱,反应按非环状过渡态进行。(Tetrahedron Lett. 1983, 24, 2865.)
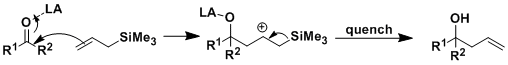
- 反应实例
Denmark等人、开发的用手性lewis碱实现了不对称烯丙基化。[1]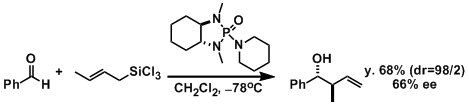
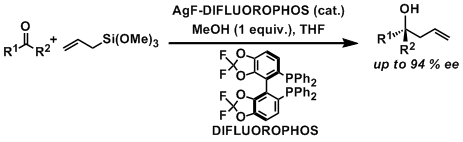
山本等人、用氟化银基手性不对称膦配体,将反应活性差,选择性低的酮实现对映选择性的烯丙基化。[2]
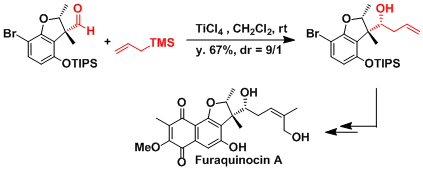
Furaquinocin A的合成[3]
Halichlorine的合成[4]
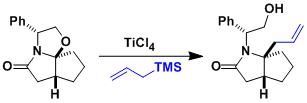
环状氧鎓盐的细见-樱井反应以高立体选择性进行。6元环的4位取代基及五元环的3位取代基对烯丙基化的作用较大。[5]
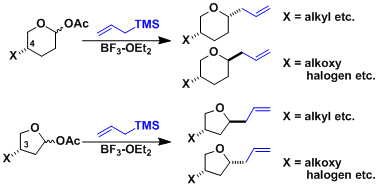
- 实验步骤
不饱和酮的共轭烯丙基化[6]
- 实验技巧
- 参考文献
[2] Wadamoto, M.; Yamamoto, H. J. Am. Chem. Soc. 2005, 127, 14556. DOI: 10.1021/ja0553351
[3] Trost, B. M.; Thiel, O. R.; Tsui, H.-C. J. Am. Chem. Soc. 2003, 125, 13155. DOI: 10.1021/ja0364118
[4] (a) Trauner, D.; Schwartz, J. B.; Danishefsky, S. J. Angew. Chem. Int. Ed. 1999, 38, 3542.[abstract] (b) Trauner, D.; Danishefsky, S. J. Tetahedron Lett. 1999, 40, 6513. doi:10.1016/S0040-4039(99)01170-3
[5] (a) Woerpel, K. A. et al. J. Am. Chem. Soc. 1999, 121. 12208. DOI: 10.1021/ja993349z (b) Woerpel, K. A. et al. J. Am. Chem. Soc. 2003, 125, 14149. DOI: 10.1021/ja0375176 (c) Woerpel, K. A. et al. J. Am. Chem. Soc. 2003, 125, 15521. DOI: 10.1021/ja037935a (d) Woerpel, K. A. et al. J. Am. Chem. Soc. 2005, 127, 10879. DOI: 10.1021/ja0524043
[6] Org. Synth. 1984, 62, 84.
概要
二异丁基氢化铝(Diisobutylaluminium hydride: DIBAL, DIBAL-H)是化学式为HAliBu2的一种还原剂。以二聚体的形式存在。
铝因其核外电子空轨道使它具有路易斯酸性。因为这一特性、使它与氢化铝锂(LAH)等氢负离子型还原剂有不同的用途和适用性。
可将腈还原为亚胺、通过水解转变为相应醛。因此,腈类化合物也被当做醛的等价体。
缩醛也可由它还原为醚。例如,亚苄基缩醛(或甲氧基苄基缩醛)经DIBAL处理后,缩醛上立体位阻小的氧原子作用后,使单侧碳氧键断裂开环生成醚。(参考PMB保护)
若用DIBAL还原酯,低温下进行虽可以部分将酯还原成醛,但一般不那么容易。加2当量以上的DIBAL将酯还原到醇,然后再氧化成醛,这样可行但步骤稍繁琐。例外,五元或六元环状内酯的话,部分还原却容易进行。近年,通过给氢负离子还原剂中添加NaOtBu可实现部分还原。
DIBAL对炔烃还原时会发生氢金属化加成。
加入n-BuLi后生成氢负离子型还原剂LiAlH(i-Bu)2(n-Bu)、是与DIBAL不同还原形式的更强的还原剂。
基本文献
・Ziegler, K., Martin, H.; Krupp, F. Ann. 1960, 629, 14. DOI: 10.1002/jlac.19606290103.
反应实例
DIBAL对炔烃还原时氢金属化加成实例。
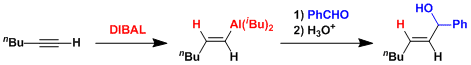
DIBAL+NaOtBu(SDBBA)、可在冰浴冷却的简便条件下将酯还原为醛。[1]
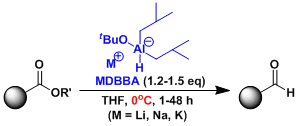
实验步骤
实验技巧
参考文献
[1] Song, J. I.; An, D. K. Chem. Lett. 2007, 36, 886. doi:10.1246/cl.2007.886
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
芳香族化合物 —> 芳香酮
- 概要
在路易斯酸存在的条件下,让酰卤或者酸酐作为酰化剂,与芳香化合物进行的酰化反应。供电子基团取代的芳香环相比于吸电子基团取代的芳香环反应性更高,更容易反应。
由于傅-克烷基化反应常常会生成重排以及多取代的副产物,所以除了分子内的烷化反应以外一般不常使用。但是,傅-克酰基化反应是吸电子取代反应,所以反应通常会停留在第一阶段。所以相对于烷基化反应,傅-克酰基化也常常被应用于分子间的反应。
- 基本文献
・Friedel, C.; Crafts, J. M. Compt. Rend. 1877, 84, 1450.
・Crafts, J. M.; Ador, E. Ber. 1877, 10, 2173.
・Crafts, J. M.; Ador, E. Bull. Soc. Chim. France 1880, 531.
・Price, C.C. Org. React. 1946, 3, 1.
・Gore, P. Chem. Rev. 1955, 55, 229. DOI: 10.1021/cr50002a001
・Groves, J. K. Chem. Soc. Rev. 1972, 1, 73. DOI: 10.1039/CS9720100073
・Eyley, S. C. Comprehensive Organic Synthesis 1991, 2, 707.
・Heaney, H. Comprehensive Organic Synthesis 1991, 2, 733.
- 反应机理
以亲电芳香环取代反应(SEAr)的形式进行的反应。首先酰卤与三氯化铝,形成强吸电子的酰阳离子,作为活性很高的中间体进行后续的酰化反应。
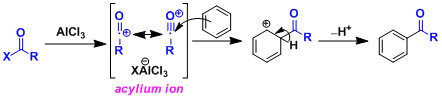
- 反应实例
由于傅-克烷基化反应无法停留在第一阶段而只生成单取代的产物,所以通常,首先利用傅-克酰基化反应酰化后,再利用克莱门森(Clemmensen)还原等,最终得到单取代的烷基化产物。
以下是给出的一个实例[1]
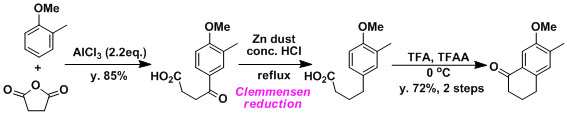
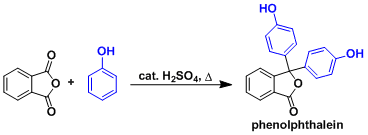
酚酞的合成
- 实验步骤
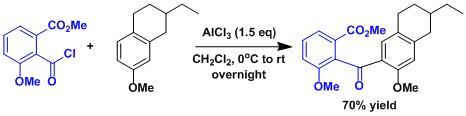
苯甲醚衍生物(380 mg, 2 mmol)首先溶解在二氯甲烷(5 mL)中,在氮气保护下冷却到0℃。缓慢加入无水三氯化铝(400 mg, 3 mmol),混合搅拌15分钟。然后在0℃缓慢滴加酰氯(456mg, 2 mmol)的二氯甲烷溶液(5 mL),搅拌30分钟后,升温到室温,搅拌过夜。反应结束后,把混合物倒入含有浓盐酸(2mL)的冰水溶液中(5mL)。接着搅拌10分钟后,用二氯甲烷(20 mL)分三次萃取。收集有机相,再用水和饱和食盐水萃取干净后,用硫酸钠干燥完毕。最后滤掉干燥剂,减压真空抽干得到的粗产物再用硅胶柱层析法(石油醚/乙酸乙酯=4/1)分离纯化得到半固体状的酮(535mg, 收率70%) [2]
- 实验技巧
※猝灭的时候,一定要把反应液缓慢的倒入冰水混合溶液中。绝对不能把冰水混合溶液倒入反应液中。如果这样做的话,水会和三氯化铝剧烈反应发热,可能会引起事故。
- 参考文献