作者:杉杉
导读:
近日,东华大学的刘为萍与赵圣印课题组在Angew. Chem. Int. Ed.中发表论文,共同报道一种全新的采用锰催化剂促进的二级醇、一级醇与甲醇之间的化学选择性三组分偶联反应方法学,进而成功完成一系列β,β-甲基化/烷基化二级醇分子的构建。
Manganese-Catalyzed Chemoselective Coupling of Secondary Alcohols, Primary Alcohols and Methanol
J.Tang, J.He, S. Zhao, W. Liu, Angew. Chem. Int. Ed. 2023, ASAP. doi: 10.1002/anie.202215882.

正文:
近年来,通过不同醇分子参与的催化偶联反应方法学的相关研究,已经备受有机合成化学家的广泛关注 (Figure 1a)[1]-[3]。然而,对于二级醇、一级醇与甲醇之间的三组分偶联反应方法学的相关研究,至今仍面临诸多的挑战 (Figure 1b) [3]-[5]。这里,东华大学的刘为萍与赵圣印团队共同设计出一种全新的全新的采用锰催化剂促进的二级醇、一级醇与甲醇之间的化学选择性三组分偶联反应方法学 (Figure 1c)。

首先,作者采用1-苯基乙醇1a、苯甲醇2a与甲醇作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用Mn-1作为催化剂,tBuOK作为碱,1,4-二氧六环作为反应溶剂,反应温度为130 oC,最终获得89%收率的偶联产物3aa。

在上述的最佳反应条件下,作者分别对一系列一级醇底物 (Scheme 1)以及二级醇底物 (Scheme 2)的应用范围进行深入研究。




接下来,作者通过一系列实验,对上述化学选择性偶联过程的反应机理进行进一步研究 (Figure 2)。


基于上述的实验研究以及前期相关的文献报道[3], [6],作者提出如下合理的反应机理 (Scheme 3)。

总结:东华大学的刘为萍与赵圣印团队共同设计出一种全新的采用锰催化剂促进的二级醇、一级醇与甲醇之间的化学选择性三组分偶联反应方法学,进而成功完成一系列β,β-甲基化/烷基化二级醇分子的构建。这一全新的三组分偶联反应策略具有广泛的底物应用范围、优良的官能团兼容性以及优良的化学选择性等优势。
参考文献:
- [1] K. Wang, L. Zhang, W. Tang, H. Sun, D. Xue, M. Lei, J. Xiao, C. Wang, Angew.
- Chem. Int. Ed. 2020, 59, 11408. doi:10.1002/anie.202003104.
- [2] S. Chakraborty, P. Daw, Y. Ben David, D. Milstein, ACS Catal. 2018, 8, 10300. doi:10.1021/acscatal.8b03720.
- [3] M. Schlagbauer, F. Kallmeier, T. Irrgang, R. Kempe, Angew. Chem. Int. Ed. 2020, 59, 1485. doi:10.1002/anie.201912055.
- [4] Y. Li, H. Li, H. Junge, M. Beller, Chem. Commun. 2014, 50, 14991. doi:10.1039/C4CC06933A.
- [5] A. Kaithal, L. L. Gracia, C. Camp, E. A. Quadrelli, W. Leitner, J. Am. Chem. Soc. 2019, 141, 17487. doi:10.1021/jacs.9b08832.
- [6] F. Freitag, T. Irrgang, R. Kempe, J. Am. Chem. Soc. 2019, 141, 11677. doi:10.1021/jacs.9b05024.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
本文作者孙苏赟
一、碘代反应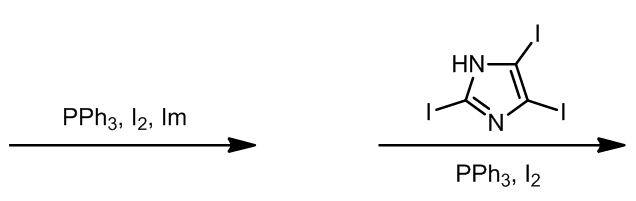

这种组合是最常用的碘代的方法,例如[1]:

此方法也常用于糖化学中:

此外还有一些不太常用的方法:

二、磺化反应
在有机合成中,磺酸酯作为效果还不错的离去基团,在有机合成中常作为卤素原子的替代,实现一系列的转化,较常用的磺酸酯的有甲磺酸酯 (Ms),三氟甲磺酸酯 (Tf)和对苯甲磺酸酯 (Ts):

离去基团的离去能力:ROTf >> ROMs ~ ROTs ~ RI > RBr > RCl

其中,(a) 是最常用的Ts磺化的方法,但是烯丙基和三级烷基炔非常不稳定,并且活性非常高,因此比较难制备提纯得到纯净物;(b) 是Ms磺化最常用的方法;(c) 用于大位阻的醇化合物;(d) 途径中使用三氟甲磺酸酐来制备Tf磺酸酯,二级醇的反应性更好,又因为可以发生SN2,因此在糖化学中非常常用。
这里是两个例子:
(1) 邻二醇的磺化-取代反应[2]:

(2) 二级醇的反应[3]:

三、磷酸酯化反应
磷酸酯在有机合成中不是非常常用,但是有时却是非常有效的反应试剂,例如Newhouse课题组开发的环酮的脱氢反应[4]:

但是磷酸酯的合成是很简单的:

反应过程中磷的手性会发生翻转。
四、低聚核苷酸的合成(oligonucleotide synthesis) [5, 6]
在低聚核苷酸的合成研究初期,磷化合物是其研究的核心。其中一个非常主要的问题就是分子中P-O–的结构的导致反应的选择性问题。这个问题也导致了后来的磷酸基团选择性活化的问题。
然而,在此之后Marrin Carruthers开发了一种亚磷酰胺的方法可以解决以上的难题。亚磷酰胺是一种很稳定的磷酸化试剂。而到了今天,这类反应常常以固相反应的形式在硅胶上进行。其中,腺嘌呤核苷和胞嘧啶上的氨基使用苯基作为保护基,鸟嘌呤核苷上的氨基可使用异丙基保护,而胸腺嘧啶的氨基则不需要保护基。
Carruthers开发的方法基本如下:


REFERENCES
- J. Am. Chem. Soc.1999, 7423-7424, DOI: 10.1021/ja991538b
- Org. Lett.1999, 11, 71-74,DOI: 10.1021/ol9905506
- Org. Lett.1999, 13, 499-502, DOI: 10.1021/ol990102y
- Org. Lett. 2018, 20, 684–687, DOI: acs.orglett.7b03818
- Science 1985 230 281, DOI: 1126/science.3863253
- Tetrahedron 1992, 48, 2223, DOI: 1016/S0040-4020(01)88752-4
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者孙苏赟
一、氯代反应
(1). SOCl2或PCl3/PCl5
这是最常见的醇的氯化的方法。
反应机理:

使用氯化磷作为氯源的反应机理是与此类似的。
(2) Ph3P/NCS (or (Cl3C)2CO, or CCl4)
这个方法中生成的膦氧化物中间体是一个很好的离去基团,对于底物具有很广泛的适应性,并且烯丙基醇的反应不会发生重排过程。
反应机理:

上边的是六氯丙酮的反应机理,NCS和CCl4的机理也是类似的:

(3) MeLi then TsCl/LiCl:这个方法中使用烷基锂试剂作为碱,会形成氧负离子,适用于大位阻的醇化合物。
(4) TsCl/NaCl

这个方法对于烯丙基醇也具有优势,具有较好的区域选择性:

在这个例子中,避免了烯丙基的重排,可以使烯丙基醇单一的转化成烯丙基氯化合物:

(4) 此外还有几种相对不是很常用的方法:

使用DMS和DCS搭配,可以对烯丙醇和苄醇有很好的选择性,并且其他饱和醇类在此条件先呈现惰性。
第二个方法其实是和Mitsuobu反应类似的,相同地,反应中会导致手性中心的反转。这个方法对于烯丙基醇和饱和醇都是适用的。
两个例子:
- 这是Meyer课题组开发的一种方法,这种方法对于四取代的烯丙醇比较有效[1]。

2. 磷试剂根据实际情况可能需要进行筛选[2]:

二、溴代反应

以上的方法在一级醇上会比二级醇上的效果好一些。反应过程是通过形成溴-膦正离子物种中间体进行的:

例如:
(1) 溴代过程中实现构型翻转[3]:

(2) 在Li+的协助下,溴负离子亲核进攻完成溴代反应[4]:

REFERENCES
- J. Org. Chem.199560247796-7814,DOI: 10.1021/jo00129a021
- J. Am. Chem. Soc.2002, 124, 15196-15197, DOI:10.1021/ja028936q
- J. Org. Chem.1965, 308, 2635-2639, DOI: 10.1021/jo01019a030
- Org. Lett.1999, 14, 645-648, DOI: 10.1021/ol990723r
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者孙苏赟
概要
Mitsunobu反应可以将一级醇和二级醇的羟基转化成很多种其他的官能团,譬如胺、硫醚等,在有机合成中的应用十分广泛。

Mitsunobu反应中,三烷基膦是至关重要的试剂,可以将醇的羟基转化成为很好的离去基团,在当初此反应发明后,又有了很广泛的发展。对于亲核试剂,酸性要在pKa 10一下。当反应中加入ZnCl2或酰亚胺类化合物,反应物的构型可以实现完全的反转,若反应物是羧酸类化合物,构型的反转的酯化反应也是会发生的,这里可以将生成的酯再水解得到和反应物相反构型的羧酸化合物。此外,DIAD可以替换DEAD作为反应试剂,在反应中起到相同的作用。

反应机理[1]:

从机理上来看,PPh3先和DEAD作用,生成的中间体再和醇发生一系列质子转移过程,最后亲核试剂进攻醇的膦离子加合物,在此加合物中,具有一个很好的离去基团,亲核试剂进攻可以顺利的进行SN2,实现构型的反转,得到相反构型的化合物。
应用实例
(1) 酰胺化反应[2]:

(2) 实现不对称二级醇的构型反转[3]:

在这个反应中,先通过Mistunobu反应生成相反勾心的酯实现羟基构型的反转,再水解得到和反应物相反构型的二级醇。
(3) 叠氮取代[4]:

这个反应中使用了(PhO)2P(O)N3作为亲核试剂的来源:

(4) 实现大环的关环[5]:

除去之前所说传统的Mistunobu反应中使用的DIED和DEAD,还有一些改进的Mistubonu反应,使用的是改进的偶氮试剂和其他助剂:

例如:
(1) 烯丙基醇的反应[6]:

(2) CMBP的应用:

反应机理:

REFERENCES
- J. Org. Chem.1987, 52, 194, 235-4238 DOI:10.1021/jo00228a016
- J. Am. Chem. Soc. 1990, 112, 760, DOI: 10.1021/ja00158a040
- J. Am. Chem. Soc. 1999, 121, 5467, DOI: 10.1021/ja990404v
- J. Am. Chem. Soc.1999, 121, 33, 7702-7703,DOI: 10.1021/ja991714g
- J. Am. Chem. Soc. 2003, 125, 27, 8112-8113, DOI: 10.1021/ja036011k
- Trahedron Lett., 1994 35 5081 DOI: 1016/S0040-4039(00)73326-0
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
- 概要
最常用的还原剂之一。对空气中的水气和氧较稳定,操作处理容易。也能推广适用于工业规模, 因为溶解性的问题,通常使用甲醇、乙醇作为溶剂。
特别是将酮,醛还原到醇的时候,该试剂是首选。反应通常进行的很快,而且简便易行。
通常情况下,硼氢化钠无法还原酯,酰胺,羧酸及腈类化合物,但当酯的羰基α位有杂原子存在时例外,可以将酯还原。(有可能是相邻杂原子的定位作用)。
对于α,β不饱和羰基化合物、优先发生1,4-还原、加入铈盐的话则优先发生1,2-还原(Luche還元)。
- 基本文献
・Schlesinger, H. I. et al. J. Am. Chem. Soc. 1953, 75 , 186. DOI: 10.1021/ja01097a049
・Dalla, V.; Catteau, J. P.; Pale, P. Tetrahedron Lett. 1999, 40, 5193. doi:10.1016/S0040-4039(99)01006-0
- 反应机理
- 反应实例
BF3存在下反应进行时、能将羧酸还原到醇。[1]

碘存在下、体系中可生成BH3-THF络合物,这样一来也可将羧酸还原为醇。[2]

- 实验步骤
- 实验技巧
- 参考文献
[2] McKennon, M. J.; Meyers, A. I.; Drauz, K.; Schwarm, M. J. Org. Chem. 1993, 58, 3568. DOI:10.1021/jo00065a020
- 概要
一般来说,由醇直接还原生成烷的转化较为困难。通常、将醇变为卤化物、磺酸酯等容易离去的基团后,再加入金属氢负离子源(LiAlH4、LiHBEt3、Bu3SnH+自由基引发剂等)来实现醇到烷的转化。熟为人知的将卤化物还原脱卤的经典条件是催化加氢、Birch还原等。
醇的卤化可用PX3、PX5、SOCl2、(COCl)2等简便易得的反应试剂来实现。若想要反应在中性条件下进行,可用Appel反応。醇的磺酸酯化可在MsCl-Et3N、TsCl-Py(-DMAP)体系中发生 。
脂肪族伯醇以外的醇可在LiAlH4-AlCl3(1:3)的条件下还原、但由于形成碳正离子中间体、基团迁移,异构化等较容易发生。
伯醇在NaBH3CN-(PhO)3PCH3I条件下、原位生成碘代烷然后继续还原得到烷烃。
自由基条件下脱去醇羟基,可通过Barton-McCombie脱氧化来完成。
- 基本文献
- 反应机理
可以通过氢负离子的亲核取代机理来解释。
![]()
- 反应实例
- 实验步骤
- 实验技巧
- 参考文献
醇→卤化物

- 概要
Appel Reaction能使伯醇・仲醇化合物转变成卤化物的反应。反应在中性条件下进行,所以对那些对酸碱不稳定的醇的合成非常有用。
- 基本文献
・Downie, I.; Holmes, J.; Lee, J. Chem Ind. 1966, 22, 900.
・Calzada, J. G.; Hooz, J. Org. Synth. 1974, 54, 63. [PDF]
・Appel, R. Angew. Chem. Int. Ed. 1975, 14, 801. doi:10.1002/anie.197508011
・van Kalkeren, H. A.; van Delft, F. L.; Rutjes, F. P. J. T. Pure Appl. Chem. 2013, 85, 817. doi:10.1351/PAC-CON-12-06-13
- 反应机理
本反应充分利用磷氧键较强的作用力。

- 反应实例
底物的立体化学会反转。[1]

使用反应性高的六氯丙酮[2a]或六溴丙酮[2b]、反应在非常低的温度下也能进行。像1-金刚烷醇之类的叔醇的底物也能适用于该反应。
利用Appel反応的膦的不对称氧化[3]

近年,研究也开发了一些利用催化量磷的方法,方便后处理分离除去[4]

- 实验步骤
香叶基氯的合成[5]
- 实验技巧

※ 用CBr4的溴化反应在0℃~室温能进行。但利用CCl4的氯化需要在加热或回流条件。
※实验产生的氧化膦后处理分离时会非常麻烦。甲磺酰化→卤代化合物(Finkelstein反応)的2步反应是本反应的代替法。
- 参考文献
[1] Suzuki T. et al. Tetrahedron Lett. 2001, 42, 65. doi:10.1016/S0040-4039(00)01880-3
[2] (a) Magid, M. R.; Fruchey, S.; Johnson, W. L.; Allen, T. G. J. Org. Chem. 1979, 44, 359. DOI:10.1021/jo01317a011 (b) Tongkatea, P.; Pluempanupata, W.; Chavasiri, W. Tetrahedron Lett. 2008,49, 1146. doi:10.1016/j.tetlet.2007.12.061
[3] Bergin, E.; O’Connor, C. T.; Robinson, S. B.; McGarrigle, E. B.; O’Mahony, C. P.; Gilheany, D. G. J. Am. Chem. Soc. 2007, 129, 9566. DOI:10.1021/ja072925l
[4] (a) Denton, R.; An, J.; Adeniran, B.; Blake, A.; Lewis, W.; Poulton, A. J. Org. Chem. 2011, 76, 6749. doi:10.1021/jo201085r (b) van Kalkeren, H. A.; Leenders, S. H. A. M.; Hommersom, C. A.; Rutjes, F. P. J. T.; van Delft, F. L. Chem. Eur. J. 2011, 17, 11290. DOI: 10.1002/chem.201101563
[5] Calzada, J. G.; Hooz, J. Org. Synth. 1974, 54, 63. [PDF]
- 相关书籍
- 相关链接
・Appel Reaction(Wikipedia)
- 概要
用二甲亚砜(DMSO)-乙二酰氯(草酰氯)体系对醇的氧化称为斯文氧化。
DCC(Pfitzner-Moffatt氧化)、TFAA(Swern变换)、无水醋酸(Albright-Goldmannyang氧化)、SO3-吡啶(Parrikh-Doering氧化)中都用到DMSO作为活化试剂,但与乙二酰氯的氧化体系副反应较少。最初用TFAA的方法活性非常高,但副反应也较多。
反应后试剂产生的副产物沸点低,容易除去,常用于大量合成醛,但是,反应会生成化学当量的二甲硫醚,有恶臭的气味。
- 基本文献
・Huang, S.-L.; Omura, K.; Swern, D. J. Org. Chem. 1976, 41, 3329. DOI: 10.1021/jo00882a030
・Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2489. doi:10.1021/jo00406a041
・Omura, K.; Swern, D. Tetrahedron 1978, 34, 1651. doi:10.1016/0040-4020(78)80197-5
・Review: Mancuso, A. J.; Swern, D. Synthesis 1981, 165. doi:10.1055/s-1981-29377
・Marx, M.; Tidwell, T. T. J. Org. Chem. 1984, 49, 788. DOI: 10.1021/jo00179a009
・Review: Tidwell, T. T. Org. React. 1990, 39, 297.
・Review: Tidwell, T. T. Synthesis 1990, 857. doi:10.1055/s-1990-27036
・Review: Lee, T. V. Comprehensive Organic Synthesis 1991, 7, 291.
- 反应机理
DMSO与乙二酰氯反应,生成氯锍盐中间体作为氧化活性剂,这一试剂对水敏感,且温度超过-60°C迅速分解,所以,此反应必须在低温(-78°C)及无水条件下进行。另外,有毒气体CO和恶臭气味的Me2S的生成,反应须在通风橱里进行,这样,此反应的缺点是其操作较复杂。

副反应中最常见的是甲硫甲基醚化(MTM化)。温度高时反应中的氯锍盐发生Pummerer异构的产物与醇反应,生成MTM保护产物。

- 反应实例
温和的条件下进行,可用于不稳定的醛的合成。是多数氧化反应中最常用的反应。以下是具体合成(+)-Thiazinotrienomycin E的实例[1]

TFAA条件的实例[2]

- 实验步骤

乙二酰氯(2.1 mL, 24 mmol)的二氯甲烷溶液(30 mL) 冷却到-78°C ,滴加DMSO(3.3 mL, 21 mmol)的二氯甲烷溶液(32 mL)。 迅速有大量气体产生,5分钟后加入醇 (4.0 g, 20 mmol)的二氯甲烷(26 mL),-78°C搅拌15分钟。然后一次性加入三乙胺(14 .0 mL, 100 mmol), -78 °C下搅拌10分钟后,缓缓升至室温,用二氯甲烷稀释后,饱和氯化铵水溶液、食盐水(2次)洗涤,无水硫酸镁干燥,过滤后溶媒在通风橱内减压蒸去(有恶臭气味),flash柱色谱(石油醚/乙酸乙酯 = 9:1)纯化精制得到目标产物醛为无色液体(3.88 g,96%)。[3]
- 实验技巧
- 通常用二氯甲烷做溶剂,此外THF、乙醚也能使用
- 除非有特别要求,一般都是以底物:乙二酰氯:DMSO:三乙胺 = 1:2:3:6的摩尔比作为标准配比来进行反应。
- DMSO产生的恶臭气味,全部操作都要在通风橱内,使用后的玻璃仪器用次氯酸水溶液(bleach)浸泡,能除去恶臭的气味。
- 参考文献
[2] Braish, T. F.; Saddler, J. C.; Fuchs, P. L. J. Org. Chem. 1988, 53, 3647. DOI: 10.1021/jo00251a001
[3] Taillier, C.; Gille, B.; Bellosta, V.; Cossy, J. J. Org. Chem. 2005, 70, 2097. DOI: 10.1021/jo048115z
- 相关书籍
醇→醚
- 概要
苄基保护基对酸性,碱性条件稳定,是普遍适用性较高的保护基。
在Brønsted酸的存在下,用BnO(=NH)CCl3,可得到在碱性条件下不稳定的苄基醚。
脱保护时一般需在还原条件下(H2-Pd/C、Na/NH3电解还原等)进行。用RuCl3/NaIO4氧化到醇,溴代硼烷等lewis酸+亲核剂的条件也能脱保护。
- 基本文献
・Czernecki, S.; Georgoulis, C.; Provelenghiou, C. Tetrahedron Lett. 1976, 17, 3535. DOI:10.1016/S0040-4039(00)71351-7
・White, J. D.; Reddy, G. N.; Spessard, G. O. J. Am. Chem. Soc. 1988, 110, 1624. DOI:10.1021/ja00213a047
- 反应机理
1. 保护
与Williamson醚合成同样的机理进行

2. 脱保护(还原条件)
- 反应实例
保护·脱保护的典型实例[1]:苄基较稳定,通常在合成的初期阶段导入

O-Bn共存下,N-Bn的选择脱保护[2]

Dubley试剂的苄基化[3]:试剂稳定,操作处理容易,中性条件下反应能进行。

- 实验步骤
NaH存在下,DMF中与BnCl或BnBr作用高收率的生成保护体。
加入催化剂量的Bu4NI(TBAI)或NaI,会促进保护反应更充分进行。(体系中的BnCl或BnBr转变为反应活性高的BnI)
- 实验技巧
溴苄刺激眼睛是催泪物质,操作时应在通风橱中进行。
淬灭终止反应时,不要加水,加入碳酸钾-甲醇并搅拌一阵,会使催泪的溴苄分解,使过滤和接下来的后处理简洁。
- 参考文献
[2] Kroutil, J.; Tmka, T.; Cemy, M. Synthesis 2004, 446. DOI: 10.1055/s-2004-815937
[3] (a) Poon, K. W. C.; Dudley, G. B. J. Org. Chem. 2006, 71, 3923. DOI: 10.1021/jo0602773 (b) Poon, K. W. C.; House, S. E.; Dudley, G. B. Synlett 2005, 3142. DOI: 10.1055/s-2005-921898
醇→乙醚
- 概要
p-甲氧基苄基(PMB or MPM)和苄基一样能对羟基进行保护・脱保护。可以利用DDQ(二氯二氰对苯醌)在温和氧化条件下,或在强酸性条件下使其脱去。
二甲氧基苄基有两种,甲氧基分别在2,4位(DMB)或3,4位(DMPM)。(DMB or DMPM)比PMB能在更温和的条件下进行脱保护。
PMB三氯乙酰亚胺(PMB-O(C=NH)CCl3)可以使PMB在酸性条件下保护。对碱不稳定的化合物也适用。
- 基本文献
・Marco, J. L.; Hueso-Rodriguez, J. A. Tetrahedron Lett. 1988, 29, 2459.
・Horita, K.; Abe, R.; Yonemitsu, O. Tetrahedron Lett. 1988, 29, 4139.
・Horita, K.; Yoshioka, T.; Tanaka, T.; Oikawa, Y. Yonemitsu, O. Tetrahedron 1986, 42, 3021.
- 反应机理
1.保护
本反应机理和Williamson合成反应相同

2.脱保护(DDQ)
形成的电荷转移配合物使反应物氧化、然后通过水解完成脱保护。

- 反应实例
DIBAL还原亚苄基缩醛(或甲氧基苄基缩醛)时,与缩醛上立体位阻小的氧原子作用后,使单侧碳氧键断裂开环。

PMB(Bn)基的迁移:利用DIBAL引起的亚苄基缩醛的开环反应,能使PMB(Bn)基转移到位阻大的羟基上。

苯连接的甲氧基越多,脱保护越简单。在PMB的存在下,可以有选择的脱去DMB 。[1]

(+)-Saxtoxin的合成[2]:利用PMB的给電子性,能减缓磺酰胺的吸电子性、而稳定下列的反应。

- 实验步骤
- 实验技巧
- 参考文献
[2] (a) Fleming, J. J.; Du Bois, J. J. Am. Chem. Soc. 2006, 128, 3926.DOI: 10.1021/ja0608545 (b) Fleming, J. J.; McReynolds, M. D.; Du Bois, J. J. Am. Chem. Soc. 2007, 129, 9964. DOI:10.1021/ja071501o
- 相关书籍
- 相关链接
・保護基 (Wikipedia日本)
・Protecting Group(Wikipedia)
・Protecting Group
・Stability of Protecting Group: Hydroxyl (organic-chemistry.org)
・Protective Groups (A.Myers’ Group:PDF)